Engineering Materials and Their Applications
Effects of Size and Crystallinity of CaCu3Fe4O12 on Catalytic Activity for Oxygen Evolution Reaction
2020 Volume 61 Issue 8 Pages 1698-1702

Details
2020 Volume 61 Issue 8 Pages 1698-1702
Effects of size and crystallinity of a quadruple perovskite oxide, CaCu3Fe4O12, were investigated for its catalytic activity for the oxygen evolution reaction (OER). Pristine CaCu3Fe4O12 powder sample was synthesized under high-pressure and high-temperature conditions of 8 GPa and 1000°C, and a portion was treated by ball milling for 120 min to obtain a powder sample with smaller particle size. The specific surface area significantly increased from 0.38 to 10.30 m2g−1 by ball-milling, leading to increased OER activity. However, it was found that the milling did not improve the OER activity efficiency in proportion to that expected from the increase in specific surface area as determined by Brunauer-Emmett-Teller analysis of adsorption/desorption isotherms measured with nitrogen gas because the crystallinity was lowered by ball milling, which suppressed the catalytic activity. This study provides important information on how to achieve the best OER catalytic performance in terms of both size and crystallinity.

The oxygen evolution reaction (OER) is an important electrochemical reaction for water splitting, industrial electrolytic anodes, and rechargeable metal-air batteries.1–3) A large overpotential for the OER results in energy loss, side reactions, and degradation of electrodes. Although noble metal oxides such as RuO2 and IrO2 have been used as catalysts to achieve a lowering of the overpotential,4–7) alternative catalysts mainly consisting of earth-abundant elements are desired because of the high cost of noble metals. Therefore, tremendous efforts have been devoted to find a means of predicting the catalytic activity for the OER including oxidation enthalpy from lower to higher oxides,1) eg electron number of transition metal ions in perovskite oxides,2) and O 2p band center relative to the Fermi level.8) In a previous study, we investigated electrochemical catalysis9–21) by perovskite oxides,9,10,13–15) quadruple perovskite oxides,15,17,19,21) mixed-anion compounds,16) pyrochlore oxides,18) spinel oxides,20) and composite sulfides.22) Among these catalyst candidates, a quadruple perovskite oxide, CaCu3Fe4O12, is one of the best OER catalysts mainly because of its unique structure and high-valent Fe4+ ions.21,23)
We also demonstrated that charge-transfer energy (Δ), which is defined by the difference between the unoccupied 3d band center (ε3d-un) of a transition metal and the 2p band center (ε2p) of oxygen, is a good descriptor of catalytic activity,11,14,15) as also reported by Hong et al.24) and Li et al.25) Among oxides with the same or similar structure, the catalytic activity, specifically the overpotential for the OER (ηOER), can be explained by Δ; the ηOER becomes smaller with a decrease in Δ. However, the actual catalytic activity is not only reflected by the intrinsic activity of a catalyst but also by many other factors such as specific surface area, crystallinity, stability, and the dispersion state of a catalyst. Therefore, detailed study of the conditions of post-synthesis treatment is required to optimize the OER catalytic performance, and in this study, we examined the effects of size and crystallinity of CaCu3Fe4O12, one of the best catalysts for the OER, using ball milling.
Pristine CaCu3Fe4O12 powder (0.2 g) obtained from a precursor consisting of Ca, Cu, Fe, and O by high-pressure synthesis at 8 GPa and 1000°C21) was pulverized by ball milling with balls 4 mm in diameter (100 g) in a container (50 ml) made of Co-based cemented carbide alloy (D40: 90 mass%WC–10 mass% Co) with the addition of 10 ml of isopropyl alcohol (IPA). The density of the Co-based cemented carbide alloy (∼14.55 g cm−3) is generally larger than those of ceramics such as ZrO2 (5.68 g cm−3), resulting in rapid pulverization, which is favorable for practical applications. The container had a double wall, and the inner wall was cooled by iced water in the outer container during ball milling. After the ball milling, some Co/WC contaminants were removed by using a magnet. The specific surface area of a sample was evaluated by Brunauer-Emmett-Teller (BET) analysis of adsorption/desorption isotherms measured with nitrogen gas using a surface area and pore size distribution analyzer (TriStarII 3020, Micromeritics Instrument Corp.). The structure of the materials was analyzed by a Rigaku UltimalV/285/DX/MO X-ray diffractometer equipped with a Mo Kα source (λ = 0.707 Å). The morphology and chemical composition of the products were characterized by field emission scanning electron microscopy (FE-SEM; S-4800, Hitachi High-Tech Co.).
Catalyst ink was prepared by dispersing a catalyst, acetylene black as a conducting agent, and polytetrafluoroethylene (PTFE) as a binder at a mass ratio of 30:3:1 in IPA for at least 60 min. For linear sweep voltammetry, the same amount (1 µg) of the catalyst composite was loaded on a uniform exposed part (10 × 10 × 1.6 mm) of a porous Ni metal electrode (Celmet®, Sumitomo Electric Industries, Ltd.) with paraffin coating by dipping the electrode in the catalyst ink several times. Linear sweep voltammetry was conducted at 1 mV s−1 and room temperature in 6 M KOH aqueous solution (pH = 15.4). All the potentials measured with an Ag/AgCl (sat. KCl) reference electrode were converted to the values versus reversible hydrogen electrode (RHE) using the following equation: E (V vs. RHE) = E (V vs. Ag/AgCl) + 0.197 + 0.05916 pH. Pt wire was used as the counter electrode.
All the pristine and ball-milled CaCu3Fe4O12 samples were confirmed to be crystallized in the quadruple perovskite structure, and the Bragg reflections in their X-ray diffraction (XRD) patterns were indexed with the CaCu3Fe4O12 structural data reported previously,21) as shown later. Figure 1 shows SEM images of the pristine CaCu3Fe4O12 powder and CaCu3Fe4O12 powder after ball milling for 10–120 min. The pristine CaCu3Fe4O12 powder consists of large polycrystals with dispersed particle size up to 10 µm because of high-temperature treatment at 1000°C and high-pressure synthesis conditions; however, control of the size at high temperature is difficult due to the phase stability of the quadruple perovskite. The pristine polycrystals were efficiently pulverized into fine powders by increasing the milling time. Since the particle sizes were inhomogeneous and had a size distribution even after ball milling, the specific surface areas were quantitatively evaluated by BET analysis of adsorption/desorption isotherms measured with nitrogen gas as listed in Table 1. As can be seen in Table 1, the specific surface area increased with the milling time from 0.38 (pristine, 0 min) to 10.30 m2g−1 (120 min). To evaluate the catalytic activity of CaCu3Fe4O12 powders with different milling time, a Ni metal electrode (Celmet®) was modified with the CaCu3Fe4O12 powders. Figure 2 shows images of the Ni metal electrode before and after modification with CaCu3Fe4O12 powder after milling for 120 min, indicating that the CaCu3Fe4O12 powder was well dispersed on the surface of the electrode.

FE-SEM images of (a) pristine CaCu3Fe4O12 powder, and CaCu3Fe4O12 powder after ball milling for (b) 10 min, (c) 30 min, (d) 60 min, and (e) 120 min.


Photographs of porous Ni metal electrode (Celmet) (a) without and (b) with CaCu3Fe4O12 after ball milling for 120 min. FE-SEM images of the porous Ni metal electrode (c) without and (d) with CaCu3Fe4O12 after ball milling for 120 min.
Figure 3(a) shows the linear sweep voltammograms measured using the porous Ni electrodes modified with the different CaCu3Fe4O12 powders in concentrated 6 M KOH aqueous solution (pH 15.4), and the current densities at 1.55 V vs. RHE are shown in Fig. 3(b) for comparison. As can be seen in Fig. 3, higher current density is observed on the surface of all the electrodes modified with CaCu3Fe4O12 powders compared to those of the bare electrode and an electrode modified with RuO2, which is consistent with the result using dilute 0.1 M KOH aqueous solution.21) The current density was standardized by the geometric surface area of the electrode, and therefore, a larger current density is expected for a catalyst powder with larger specific surface area. As expected, the pristine CaCu3Fe4O12 powder before ball milling has the lowest current density because it has the lowest specific surface area (see Table 1) except for the electrode without catalyst and with the reference RuO2 catalyst. However, as can be seen in Fig. 3(b), the CaCu3Fe4O12 powder after milling for 10 min exhibited the highest current density in spite of its low specific surface area 2.70 m2 g−1, and the current density decreases with an increase in milling time. This observation shows that the OER catalytic activity is not solely affected by the surface area of the catalyst.

(a) Linear sweep voltammograms measured at 1 mV s−1 in 6 M KOH aqueous solution using porous Ni electrodes modified with CaCu3Fe4O12 powders after ball milling for 10, 30, 60, and 120 min. Voltammograms measured with a bare porous Ni electrode and Ni electrode modified with RuO2 are also shown for comparison. Current density was standardized by the geometric surface area of the electrode. (b) Current densities at 1.55 V vs. RHE in the voltammograms.
Figure 4 displays the XRD profiles of the CaCu3Fe4O12 powders before and after ball milling. Although slight contamination with Co/WC from the balls and container is seen, it was confirmed that the contamination does not affect the catalytic activity in the presence of CaCu3Fe4O12 in our preliminary experiment. Table 2 summarizes the position and full width at the half maximum (FWHM) of the 220 peaks of the XRD profiles shown in Fig. 4. The 220 peak position is monotonically shifted from the original position at 15.90° to a lower angle as milling time increases, indicating an increase in the lattice constant (3.6% increase after milling for 60 min). The increase in the lattice constant implies a decrease in Fe valence (increase in Fe ionic radius) resulted from the introduction of oxygen vacancies. However, further detailed investigation should be required to obtain quantitative information on the valence change. In addition, the FWHM monotonically increases with milling time from 0.254° to 0.345°. These results suggest that the crystallinity of CaCu3Fe4O12 powder is degraded with introduction of defects by ball milling.

XRD patterns of pristine CaCu3Fe4O12 sample, and CaCu3Fe4O12 powders after ball milling for 10, 30, and 60 min.

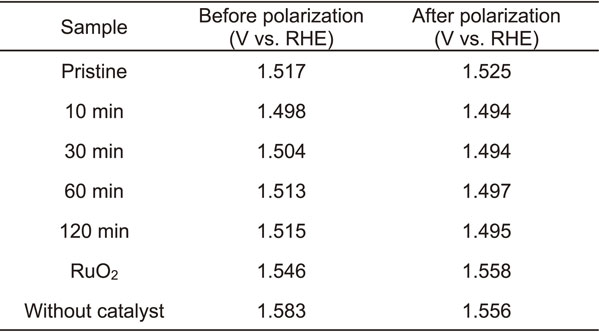
The stability of the electrodes was evaluated by chronoamperometry at 1.6 V vs. RHE in 6 M KOH aqueous solution, as displayed in Fig. 5. The order of the current density curves was almost the same as that for the voltammograms shown in Fig. 3. The current density increased with modification of the CaCu3Fe4O12 powder by ball milling, but the largest current density was attained after ball milling for 10 and 30 min. After a small initial increase in current density as the potential was held at 1.6 V, the current density slightly declined but remained almost constant until 6 h of holding time. In contrast, the current density gradually increased with potential holding time on the surfaces of the bare electrode and the electrode modified with RuO2, and the current density was far lower than those on the electrodes modified with CaCu3Fe4O12. The increase in the current density can possibly be attributed to the formation of an amorphous layer on the surface of the electrode, resulting in an increase in the surface area because of the concavity and convexity of this amorphous layer.21) Linear sweep voltammogram after chronoamperometry (Fig. 6) revealed that the current densities on the electrodes modified with CaCu3Fe4O12 after ball milling converged to an average curve; higher activity becomes slightly lower and lower activity becomes slightly higher, implying reconstruction and homogenization of the surface of CaCu3Fe4O12. It should be noted that the current density slightly decreased with modification of the pristine CaCu3Fe4O12 powder. The onset potentials before and after chronoamperometry listed in Table 3 also show a similar tendency; the onset potentials of electrodes modified with CaCu3Fe4O12 after milling converged to 1.50 V vs. RHE (i.e. ηOER = 0.27 V), and those of electrodes modified with pristine CaCu3Fe4O12 and RuO2 slightly increased, suggesting that surface reconstruction of the CaCu3Fe4O12 is promoted by the milling process.
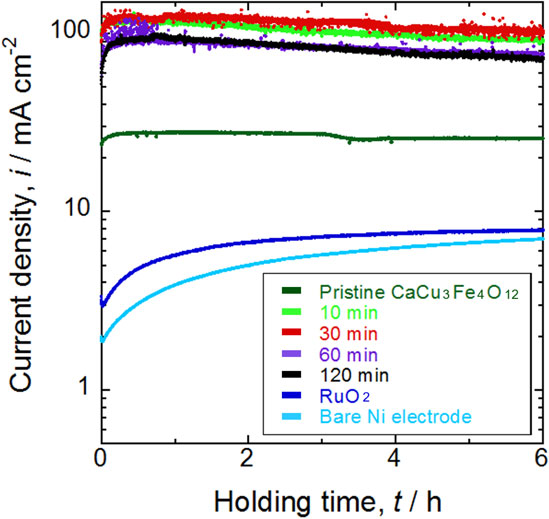
Chronoamperograms measured at 1.6 V vs. RHE in 6 M KOH aqueous solution using porous Ni electrodes modified with RuO2 or different CaCu3Fe4O12 powders after ball milling for 10, 30, 60, and 120 min. Amperograms measured with a bare porous Ni electrode and Ni electrode modified with RuO2 are also shown for comparison. Current density was standardized by the geometric surface area of the electrode.

Linear sweep voltammograms measured after chronoamperometry for 6 h at 1 mV s−1 in 6 M KOH aqueous solution using porous Ni electrodes modified with different CaCu3Fe4O12 powders after ball milling for 10, 30, 60, and 120 min. Voltammograms measured with a bare porous Ni electrode and Ni electrode modified with RuO2 are also shown for comparison. Current density was standardized by the geometric surface area of the electrode.
The effects of size and crystallinity on electrochemical catalytic activity were investigated using CaCu3Fe4O12 powder as a model catalyst. The size of CaCu3Fe4O12 particles was successfully reduced by ball milling using cemented carbide alloy in isopropyl alcohol. As can be expected, the size reduction by ball milling enhanced the current for the OER, but longer milling degraded the crystallinity of CaCu3Fe4O12, resulting in inferior catalytic performance. The defects introduced by milling promoted surface reconstruction of the catalyst, and catalytic activity of the milled CaCu3Fe4O12 converged to the same performance after 6 h of polarization at 1.6 V vs. RHE in 6 M KOH aqueous solution. In conclusion, size reduction of catalysts is favorable for the OER, but destruction of the catalyst structure should be avoided. We are now investigating methods to reduce the size of catalysts without destruction of their structure by mixing easily removable impurities during the synthesis process, in addition to large-scale synthesis using large-volume high-pressure equipment for further tests as practical electrocatalysts.
This research was supported by a Grant-in-Aid for The University of Tokyo Excellent Young Researcher and Grant-in-Aid for Scientific Research (17K18973, 17K19182, 18H03835, 19H02438, 20H02825, and 20H05180) commissioned by the Japan Society for the Promotion of Science.