Abstract
Unsupervised machine learning (ML) is examined for the result of molecular dynamics (MD) simulation to extract characteristics of catalytic reaction. O–H bond dissociation of ethanol on Fe–Co nanoparticle in ab initio MD simulation [S. Fukuhara et al., Chem. Phys. Lett. 731 (2019) 136619] is employed as an example. Hierarchical clustering of radial distribution function successfully classifies coordinates on reaction in the dendrogram. Moreover, receiver operating characteristic curve reveals the distance to the farthest-neighbor atom from the target atom is a dominant descriptor for the clustering. An optimum structure of catalytic nanoparticle is predicted based on these automated analyses. This study shows a new way of post-process of results of MD simulations based on the unsupervised learning technique and it paves the way for a new possibility of ML-based materials design.
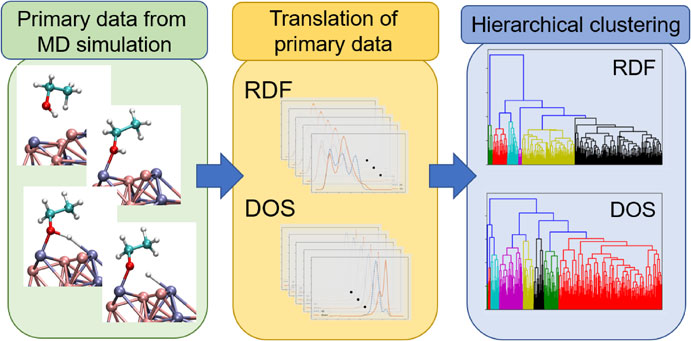
Schematic image of hierarchical clustering of structural (radial distribution function, RDF) and electronic (density of states, DOS) characteristics obtained from molecular dynamics simulation of catalytic reaction on metal nanoparticle.
Fullsize Image

