Microstructure of Materials
Effects of Local Bonding between Solute Atoms and Vacancy on Formation of Nanoclusters in Al–Mg–Si Alloys
2023 Volume 64 Issue 8 Pages 1930-1936
Details
2023 Volume 64 Issue 8 Pages 1930-1936
Nanoclusters formed in Al–Mg–Si alloys affect the aging behavior of the alloys depending on the formation temperature. In the present study, first-principles calculations were carried out to evaluate the two- and three-body interactions between Mg, Si atoms and vacancies in the Al matrix and estimate the effect of local bonding on the formation of nanoclusters. Monte Carlo simulations were subsequently performed to investigate the stable structure of the nanocluster formed in Al–0.95 mass pct Mg–0.81 mass pct Si alloy. We found that the Mg–Si and Si–Vac pairs are stable in the Al matrix. The result shows that the solute atoms easily aggregate with different types of solute atoms and that the Si atom has a strong attractive interaction with a vacancy. Furthermore, Mg–Si–vacancy triplets are more stable than Mg–Si and Si–vacancy pairs in the Al matrix. The nanoclusters in the Al matrix were thermally stabilized by the stable configurations between solute atoms and vacancy. Thus, the electronic structure calculations suggested that the local bondings within a nanocluster play a significant role in not only the thermal stability but also the formation and growth behavior of nanoclusters during aging at low temperatures.
This Paper was Originally Published in Japanese in J. JILM 72 (2022) 47–53. The abstract, the captions of Figs. 1, 2, 3, 4 and the caption of Table 3 are slightly modified.
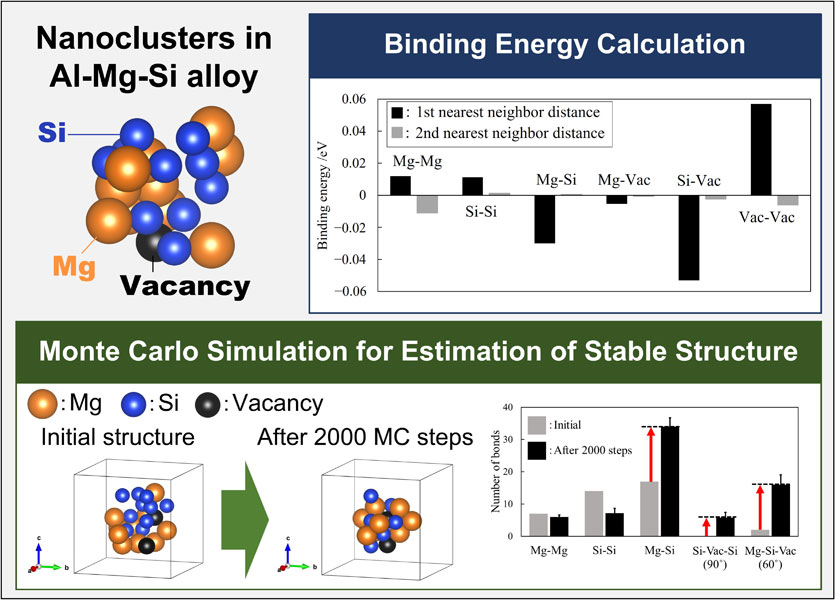
The binding energy calculations of local bonding formed in nanoclusters and Monte Carlo simulations for finding the stable structure of nanoclusters. From these results, it was found that the nanoclusters become more stable as stable atomic configurations are formed.
In recent years, automakers have been actively working to reduce the weight of car bodies using multi-materials, as a solution to environmental problems such as global warming and air pollution. The use of light metals has been progressing as car bodies become multi-materials; for instance, Al–Mg–Si alloys are often used for body panels. The advantages of using this alloy as an automotive body panels material include its excellent balance of specific strength and ductility, as well as its bake-hardenability (hereinafter referred to as BH), which increases in hardness after a short heating when the baking coating is performed. These alloys are age-hardening alloys, and their strength and elongation are significantly changed after heat treatment. Therefore, in the production line, as-quenched alloys are formed and then strength is obtained by age hardening during baking coating in the final process. However, if these alloys are held or stored at room temperature after solution treatment and quenching, not only the formability but also the BH deteriorated. As a result, the alloys cannot be obtained the expected strength. This phenomenon is believed to be caused by the complicated aging behavior of Al–Mg–Si alloys.
Aging is generally performed at a single temperature after solution treatment and quenching (one-step aging). On the other hand, aging is sometimes performed in multiple steps at different temperatures. In other words, the first stage of aging is performed at one temperature, followed by a second stage at a different temperature (usually higher than the first stage). This aging process is known as two-step aging. For example, in Al–Mg–Si alloys, pre-aging at room temperature results in a lower peak hardness at final aging than at normal aging.1) The phenomenon is called the negative effect of two-step aging, which is why the expected strength cannot be obtained when Al–Mg–Si alloys are held or stored at room temperature after solution treatment.2) On the other hand, if pre-aging is performed immediately after solution treatment and quenching at about 100°C, the negative effect is suppressed or the peak hardness is increased compared to that of one-step aging, called the positive effect of two-step aging. Such differences in aging behavior are presumed that the nanoclusters formed in the early stages of aging differ depending on the temperature conditions of pre-aging.3)
In general, aging precipitation of Al–Mg–Si alloys is considered to proceed by the following process.4)
| \begin{align} &\text{Supersaturated solid solution $\alpha$}\to \text{Nanocluster}\\ &\quad \to \text{GP zone}\to \text{$\beta''$ phase (strengthening phase)}\\ &\quad \to \text{$\beta'$ phase}\to \text{$\beta$ phase} \end{align} | (1) |
The microstructural observations using transmission electron microscopy (TEM),9,10) structural analysis using small-angle X-ray scattering11,12) and analysis using positron annihilation spectroscopy13,14) have been carried out to study the structure and composition of nanoclusters. Computer simulations have also been conducted actively. Examples of first-principles calculations studies include evaluation of the stability of metastable phases formed in Al–Mg–Si alloys by Ravi et al.,15) and Monte Carlo simulation of clustering in Al–Mg–Si alloys during early aging by Sandberg et al.16) In addition, Fallah et al. analyzed clustering of solute atoms in Al–Mg–Si alloys by combining Phase-Field and Atom Probe Tomography (APT).17) Computer simulations have also been performed to analyze precipitates in the early aging stage of alloy systems such as the Al–Cu18–21) and Al–Zn–Mg22–25) systems. On the other hand, few reports have evaluated the thermal stability of metastable phases, considering the effects of vacancies, which are considered to play an important role in forming nanoclusters in Al–Mg–Si alloys. However, unlike metastable phases, nanoclusters do not have a specific long-range structure. Therefore, thermal stability is presumed to be significantly affected by the local structure of nanoclusters. Thus, it is essential for discussing the thermal stability of nanoclusters to clarify the effect of local bonding which mainly composed of solute atoms. In this study, we focused on the bonding states between Mg, Si, and vacancies in the Al matrix, which are the elementary processes of nanocluster formation. We evaluated their binding energies by first-principles calculations. Furthermore, Monte Carlo (MC) simulations based on the Metropolis method26) were employed to explore the structure of the equilibrium state of the nanocluster model, and the effects of the local bonding of the nanoclusters on their thermal stability and formation behavior were investigated.
To calculate the total energy of the isolated state, we performed energy calculations for a model in which Al at the center was substituted by Mg, Si, and vacancies in a 256-site supercell with a 4 × 4 × 4 Al face-centered cubic lattice. Energy calculations were also performed for a similar supercell in which two Al at the center and at the first or second proximal positions were substituted with Mg, Si, and vacancies. The binding energy of the Mg, Si, and vacancy pair in Al was calculated by considering the energy difference between the isolated state and the binding state. The binding energy Eb was calculated using eq. (2).
| \begin{equation} E_{\text{b}} = (E_{\text{b}}{}^{2} + E_{\text{Al}}) - (E_{\text{s1}} - E_{\text{s2}}) \end{equation} | (2) |
To analyze the bonding state between the three solute atoms or vacancies, one atom in the central position and two atoms in the first nearest position of the central atom in a 256-site supercell of a 4 × 4 × 4 Al face-centered cubic lattice were substituted with Mg, Si, and vacancies to form a triplet. In the face-centered cubic lattice, there are four possible configurations of three atoms: three atoms adjacent at an angle of 60°, three atoms adjacent at a bond angle of 90°, 120°, and 180° between the atoms in the first nearest neighbor position. Therefore, triplet models were created by substituting Al in the matrix with Mg, Si, and vacancies in these configurations. Each arrangement is shown in Fig. 1. For models with bond angles of 90°, 120°, and 180°, some of them have several configurations, such as Mg–Mg–Vac and Mg–Vac–Mg. Binding energies between the triplet of Mg, Si, and vacancies in Al were calculated for all configurations by considering the energy difference between the isolated and bound states, respectively. The binding energy Eb was calculated by the following eq. (3). Note that Eb3 is the total energy of the supercell including the binding of solute atoms or vacancies.
| \begin{equation} E_{\text{b}} = (E_{\text{b}}{}^{3} + 2E_{\text{Al}}) - (E_{\text{s1}} - E_{\text{s2}} - E_{\text{s3}}) \end{equation} | (3) |

Atomic configurations of triplets at (a) 60°, (b) 90°, (c) 120° and (d) 180° bond angle.
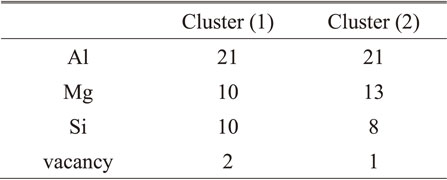
Since it is difficult to directly analyze the formation of nanoclusters in the framework of first-principles calculations from the viewpoint of computational complexity, we considered creating a model that virtually simulates the cluster structure within a limited region in the Al matrix. First, Mg, Si, and vacancies were substituted at a total of 43 atomic sites from the central atom to the third nearest neighbor atoms in the supercell with the compositions shown in Table 1, and two models simulating nanoclusters were created by randomly arranging these atoms. The Mg/Si ratio of the nanocluster model was determined based on the results of atom probe analysis. First, the median of Mg/Si ratio of Cluster (1) formed by natural aging and Cluster (2) formed by pre-aging at around 373 K were calculated. Here, about 43 atoms which is the size of the nanocluster model used in this study were used to calculate. Then, the Mg/Si ratio of Cluster (1) was set to Mg/Si = 1, which is a larger ratio of Si because Si-rich nanoclusters are considered to influence the negative effect of two-step aging. On the other hand, Cluster (2) was set to Mg/Si = 1.67, because it is reported that the ratio approaches about this ratio as it grows.6) The number of vacancies was examined by comparing the positron lifetime measurements27) of Al–Mg–Si alloys by Kani et al. with those of pure aluminum at different temperatures by Shirai.28) First, the positron lifetimes of the naturally aged and pre-aged alloys were referenced, and these values were compared to the positron lifetime measurements of pure aluminum to obtain the temperature of pure aluminum corresponding to the respective positron lifetimes. The ratio of vacancy concentration was estimated from the thermal equilibrium state at these temperatures, and it was approximated that the naturally aged material containing Cluster (1) contains about 1.67 times more vacancies than the pre-aged material containing Cluster (2). Considering that Kani et al. reported that the vacancy concentration in the matrix does not change between natural aging and pre-aged alloys and that vacancies are concentrated in nanoclusters, the modeling was conducted with the vacancies in Cluster (1):Cluster (2) = 2:1. The number of Al, Mg, Si atoms, and vacancies in the nanocluster models are shown in Table 2. The equilibrium state at each temperature was searched by MC simulations using the Metropolis method. Since tracing the formation of clusters from completely random structures is computationally expensive, stable pairs and triplets were evaluated based only on the exchange of atoms within nanoclusters.

In this study, the Vienna Ab initio Simulation Package (VASP), which performs electronic structure calculations based on density functional theory (DFT), was used.29,30)
For all calculations, the plane-wave cutoff energy of 400 eV, and generalized gradient approximation (GGA) exchange–correlation density functional by Perdew–Burke–Ernzerhof (PBE) was used.31) The k-point sampling was chosen using the Monkhorst-Pack method:32) 3 × 3 × 3 and 6 × 6 × 6 mesh partition including the Γ point, where the differences in calculation results due to the way of k-point sampling were compared to evaluate the validity.
The convergence criteria of SCF calculation were set to 1.0 × 10−5 eV, and the convergence criteria of the force on each atom was set to 0.01 eV/Å. MC simulations were performed using a 2 × 2 × 2 k-point mesh partition and 2000 steps without structural relaxation.
The binding energies of solute atoms and vacancies in Al matrix are shown in Fig. 2. Comparing of the 3 × 3 × 3 mesh partition and 6 × 6 × 6 mesh partition cases, the 3 × 3 × 3 mesh case showed lower binding energy by 8.9 × 10−3 eV at the first nearest neighbor position of Mg–Mg and 6.3 × 10−3 eV at the second nearest neighbor position, respectively. In addition, in the case of the 3 × 3 × 3 mesh partition, the binding energy was shown also lower than 6 × 6 × 6 mesh partition by 1.5 × 10−2 eV at the first nearest neighbor position of Mg–Vac and by 5.2 × 10−3 eV at the second nearest neighbor position, respectively. These results showed a tendency to underestimate the energy of supercells containing Mg atoms. This tendency was also observed in the small model with 3 × 3 × 3 Al face-centered cubic lattice and the large model with 6 × 6 × 6 Al face-centered cubic lattices in the preliminary study, it was clarified that the accuracy of k-point sampling is important, especially for Mg-containing systems. Therefore, we decided to use the results of the 6 × 6 × 6 mesh partition for the evaluation of the binding energy. The calculated binding energies indicate that the Mg–Mg and Si–Si pairs are more stable at the second nearest neighbor position than at the first nearest neighbor position, while the Mg–Si pairs is more stable at the first nearest neighbor position. These results are consistent with the tendency of interactions between solute atoms and between vacancies and solute atoms reported by Hirosawa et al.33) These results suggest that different atoms, such as Mg and Si, are more likely to form with each other than with same atoms in Al–Mg–Si alloys. In addition, since Si and vacancies are very stable at the first nearest neighbor position, there is an attractive interaction between Si and vacancies in the Al matrix, indicating that Si has the effect of trapping vacancies. It has been proposed by Yamada et al. that Si-rich clusters strongly trap vacancies,34) which is presumed to be due to the attraction between Si–Vac.

Binding energies of pair composed of solute atoms (Mg, Si) and vacancies in the Al matrix. (a) k-sampling, 3 × 3 × 3 and (b) k-sampling, 6 × 6 × 6 (Γ centered).
The binding energies between triplets composed of three solute atoms in Al are shown in Fig. 3. Note that binding energies are not listed when the structure overlaps with other triplets, such as when the Mg–Si–Mg cluster is placed at 60°. The Mg–Mg–Mg and Si–Si–Si clusters were unstable at both bond angles, whereas all triplets containing both Mg and Si were more stable than the isolated state. Thus, local structures consisting of only one type of atom without vacancies, such as Mg-only or Si-only, are unlikely to form. In addition, a comparison of binding energies of triplets containing both Mg and Si shows a tendency for the triplets to become unstable when the same type of atoms are neighboring, and to become stable when a different type of atoms are neighboring.

Binding energies of triplet composed of three solute atoms (Mg, Si) in the Al matrix.
The binding energies of solute atoms and vacancies between three-body are shown in Fig. 4. Figure 3 shows that the Mg–Mg–Mg and Si–Si–Si triplets are unstable at all bond angles. However, the triplets are stabilized when one atom is substituted by a vacancy. This tendency is more pronounced in the case of Si, which is considered to be due to the stable pairs between Si and vacancies. In particular, the Si–Vac–Si arrangement with a bond angle of 90° and the Mg–Si–Vac arrangement with a bond angle of 60° form very stable triplets, which are more stable than the Mg–Si and Si–Vac pairs. These results show that the formation of a triplets involving the vacancy may lead to the formation of a local bonding more stable than that between pairs. Although Si-rich clusters are considered to trap vacancies strongly, the existence of very stable Mg–Si–Vac 60° triplets suggests that clusters containing an equal ratio of Mg and Si (hereinafter referred to as “Mg/Si clusters”) are also effective in trapping vacancies.

Binding energies of triplet composed of two solute atoms (Mg, Si) and a vacancy in the Al matrix. (a) (Mg, Vac) and (Si, Vac) and (b) Mg, Si and Vac.
Change in energies of nanocluster models during MC simulations are shown in Fig. 5, the atomic configurations of nanoclusters before Monte Carlo simulation are shown in Fig. 6, and overall view of atomic configurations of Cluster (1) model at 298 K and Cluster (2) model at 373 K after 2000 steps Monte Carlo simulation is shown in Fig. 7, respectively. VESTA was used to draw all atomic models.35) In the MC simulation performed in this study, the stable structure was searched by repeatedly calculating the total energy by substituting the positions of atoms or vacancies for 43 atomic sites in the range from the center of the supercell to the third nearest neighbor position. And for the energy difference dE before and after the substitution, the following operation is repeated.

Change in energies of nanocluster models during Monte Carlo simulation. (a) Cluster (1) model at 298 K and (b) Cluster (2) model at 373 K.

Atomic configurations of nanoclusters before Monte Carlo simulation. (Al atoms are not represented.) (a) Cluster (1) model and (b) Cluster (2) model.

Overall view of an atomic configurations of Cluster (1) model at 298 K and Cluster (2) model at 373 K after 2000 steps Monte Carlo simulation and the cross-sectional view on a–b, b–c, and c–a planes of the model cut by parallel planes through the center of supercell. (Al atoms are not represented.)

Figure 5 shows that the total energy of both models decreased as the MC simulation progressed and approached an equilibrium state. The Cluster (1) and Cluster (2) models were simulated at two different temperature conditions (298 K and 373 K, respectively), and no clear difference in nanocluster binding was observed. The difference in cluster formation at 298 K and 373 K is assumed to be determined by the combination of the binding energy between Mg, Si, and vacancies and entropy contribution, however, the region and the composition within the region were constrained as analytical conditions in this analysis. Thus, it is considered that the difference due to temperature could not be captured in this simulation. For this reason, we did not discuss the effect of temperature on cluster formation in this analysis but focused only on the formation of pairs and triplets inside clusters. The number of Si–Vac pair, Mg–Si pair, Si–Vac–Si 90° and Mg–Si–Vac 60° triplets, which were found to be stable pairs or triplets by binding energy calculations, increased in both the Cluster (1) and Cluster (2) models compared to before the simulations.
Therefore, it was found that as these stable stable configurations are formed inside the nanocluster model, the total energy of the system decreases, and the nanocluster model becomes more stable. Since actual nanoclusters are also not composed only of solute atoms, but also contain some amount of Al, local bonding at short range, such as between two or three-body, is considered to significantly contribute to the thermal stability of the nanoclusters. Here, cross sections of the a–b, b–c, and c–a planes through the center of the nanoclusters are shown in Fig. 7 to examine in detail of the local bonding of the nanocluster models after MC simulation, in both Cluster (1) and Cluster (2) models, vacancies were found to exist near the center of the nanoclusters. This tendency was especially pronounced in the Cluster (1) model.
After the simulation, the number of stable bondings including vacancies such as Mg–Vac pair, Si–Vac pair, Si–Vac–Si 90° and Mg–Si–Vac 60° triplet increased, and vacancies existed near the center of the nanoclusters. These results indicate that the clusters are more stabilized by incorporating vacancies and vacancies are strongly trapped by the stable bondings with solute atoms such as Mg and Si once incorporated. Normally, the excess vacancies introduced during quenching disappear with decreasing temperature, and the vacancy concentration decreases. However, due to the effects of solute atoms trapping vacancies, vacancies do not disappear in the nanoclusters, and the formation of nanoclusters can maintain a higher concentration of vacancies than the thermal equilibrium concentration of vacancies in Al matrix. Table 3 shows that the number of Si–Vac–Si 90° and Mg–Si–Vac 60° triplets are larger in the Cluster (1) model, which is considered to be due to the larger number of vacancies in the Cluster (1) model, and the cohesion of the solute atoms around those vacancies. Therefore, Cluster (1) with higher vacancy concentration tends to have higher thermal stability than Cluster (2), which is presumed to be the reason why Cluster (1) is less likely to transition to the β′′ phase.
In general, Si-rich nanoclusters are considered to high thermal stability and do not recover when kept at relatively high temperatures. A higher Si ratio promotes the formation of stable configurations such as Si–Vac–Si 90° and Mg–Si–Vac 60° triplets, which explains why the thermal stability of the clusters increases with increasing Si ratio. In addition, Fig. 2 and Fig. 4 show that bondings between Si atoms are stable when they are formed through vacancies, so extremely Si-rich clusters are considered to have particularly high vacancy concentration and high thermal stability.
3.4 Effect of bonding between solute atoms and vacancies on formation and growth behavior of nanoclusterFigure 2 showed that stable pairs were formed between Mg and Si, and Si and vacancies. Therefore, in the early stage of pre-aging, many Mg–Si pairs and Si–vacancy pairs are expected to form. Aruga et al. reported that Si-rich clusters were formed in the initial stage of clustering, and later the fraction of Mg increased, and interpreted to be due to the difference in interaction energy between Si and vacancies and between Mg and vacancies.36) They also reported that Cluster (1) formed at room temperature has a larger fraction of clusters with a smaller Mg/Si ratio than Cluster (2) formed at around 373 K.37) This phenomenon can be similarly interpreted from the results of this study and can also be explained in terms of the stability of local bonding of solute atoms inside the clusters. In other words, since Si and vacancies have a strong attractive interaction, Si-rich clusters are formed in the initial stage of clustering due to the cohesion of Si against vacancies. Later, since Mg and Si form stable pairs, the Mg/Si ratio gradually increases as Mg aggregates to the initial Si-rich clusters. The rate of this process proceeding is presumed to be highly depending on the diffusion rate of atoms, and diffusion proceeds independent of temperature due to quenched vacancies in the early stages of clustering. However, when the Si-rich clusters are formed, some of vacancies are trapped in the clusters and the rate of Mg binding to the Si-rich cluster significantly depends on the temperature. If the temperature at this stage is low, Mg will not bind to the Si-rich clusters and the fraction of Si-rich clusters will remain at high. Si-rich clusters are stable with a large number of vacancies, while Mg/Si clusters with same ratio of Mg and Si can form stable Mg–Si pairs, thus Mg/Si clusters can maintain thermal stability even at lower vacancy concentrations than Si-rich clusters. For this reason, it is considered that the fraction of Si-rich clusters increases at room temperature and the fraction of Mg/Si clusters increases around 373 K. The results of MC simulation shown in Fig. 7 indicate that the nanocluster model after the simulation has a structure which solute atoms and vacancies are more concentrated than before the simulation. In addition, the total number of bondings between solute atoms and vacancies increases after the simulation compared to before the simulation. These results indicate that the nanoclusters will be more stable as the Al content in the nanoclusters decreases. Therefore, as the nanoclusters grow and bondings are formed between solute atoms and vacancies, Al atoms are carried out of the nanoclusters, and the ratio of Al atoms inside the nanoclusters is expected to decrease with nanoclusters larger growth.
In present study, to investigate the effect of bondings between solute atoms and vacancies in Al–Mg–Si alloys on the nanoclusters formed in the early aging stage, first-principles calculations were performed to determine the binding energies between two and three-body composed of Mg, Si, and vacancies in the Al matrix. Furthermore, the equilibrium structure of the nanocluster model was explored using Monte Carlo simulations, and the following findings were obtained.
This research was supported by Program on Open Innovation Platform with Enterprises, Research Institute and Academia (OPERA) (JPMJOP1843) and JSPS KAKENHI, (19H02482, 19K04993, 18H05453). This research was conducted with the supercomputer HPE SGI8600 in the Japan Atomic Energy Agency.