Abstract
In this study, we report on a case of probable sporadic Creutzfeldt-Jakob disease (sCJD) diagnosed after a difficult course of status epilepticus (SE) in a patient with poststroke epilepsy. The patient was admitted with progressive cognitive decline and convulsive SE; therefore, it was initially thought that the patient had developed SE due to nonadherence to antiseizure medication (ASM) use, but despite treatment with ASMs after admission, no improvement was noted in consciousness disturbance or lateralized periodic discharges (LPDs) on electroencephalogram (EEG) examination. After a refractory course, the progression of LPDs to generalized periodic discharges (GPDs) on EEG and abnormal magnetic resonance imaging (MRI) findings met the diagnostic criteria of sCJD. Even if the patient had epilepsy, such as poststroke epilepsy, as in this case, it is essential to consider other underlying causes, including CJD in cases of superrefractory SE.
Introduction
Creutzfeldt-Jakob disease (CJD), a type of human prion disease, is known to be a fatal neurodegenerative disorder characterized by rapidly progressive cognitive impairment and neurological symptoms such as myoclonus, which may culminate in akinetic mutism before death. There are three subtypes of human prion diseases, namely, sporadic, genetic, and acquired. The most common subtype is sporadic CJD (sCJD), accounting for approximately 85% of cases.1) Early detection of CJD symptoms is crucial to prevent transmission of this fatal disease, which has a uniformly rapid degenerative and incurable nature. While seizures may present as an early symptom of CJD in rare cases, they are generally not a common clinical manifestation.2) In this study, we observed a case of sCJD with an atypical course characterized by status epilepticus (SE) and similarities to poststroke epilepsy in clinical and electroencephalogram (EEG) findings, diagnosed after a challenging course. Herein, we report the case with a review of the literature.
Case Report
A 68-year-old woman was admitted with a severe headache and impaired consciousness. As per her head computed tomography findings, detected were aneurysmal subarachnoid hemorrhage (SAH) and intracerebral hemorrhage in the right frontal lobe. She underwent neck clipping surgery for the anterior communicating artery aneurysm at our hospital. Approximately 6 months after the onset of SAH, she experienced recurrent transient memory impairments. Based on the history and the interictal epileptiform discharges in the bilateral frontal region on EEG examination, her seizure type was focal onset impaired awareness seizure, which led to a diagnosis of poststroke epilepsy. After initiating levetiracetam, the medication compliance was good, and the symptoms disappeared for an extended period. There were almost no apparent sequelae of SAH, and cognitive function was good.
However, at the age of 78, the patient began experiencing hallucinations, abnormal behavior, and gait disturbances, with a progressive decline in cognitive function. Due to suspicion of dementia with Lewy bodies or cerebrovascular dementia, a psychiatric assessment was initiated. Concurrently, it was determined that the patient frequently did not adhere to recommended antiseizure medication (ASM) use. Three months after the initial onset of symptoms, the patient presented with disturbed consciousness. She had convulsive seizures during the EEG examination, and the EEG showed lateralized periodic discharges (LPDs) in the right hemisphere (Fig. 1-A). The convulsive seizures and EEG abnormalities promptly improved after intravenous diazepam administration. Based on the patient's poor medication adherence and the above EEG findings, she was diagnosed with convulsive SE of poststroke epilepsy. We then administered 7.5 mg/kg fosphenytoin; however, there was no improvement in consciousness, and her EEG demonstrated recurrent LPDs the following day, which led us to consider a nonconvulsive SE condition. Clinical findings remained unchanged, and LPDs persisted on EEG examination; therefore, we initiated a protocol of deep sedation and intubation with continuous EEG monitoring utilizing propofol and midazolam. Although we also began administering lacosamide (100 mg) and perampanel (2 mg) and gradually increased the dosage, reoccurrence of LPDs with extended intervals was repeatedly noted upon reducing sedation (Fig. 2). She underwent a tracheotomy on the 14th day of hospitalization due to long-term intubation. We continued searching for the cause of the acute symptomatic seizures because the superrefractory nature of the condition under high-dose ASMs led us to consider the presentation atypical as poststroke epilepsy.

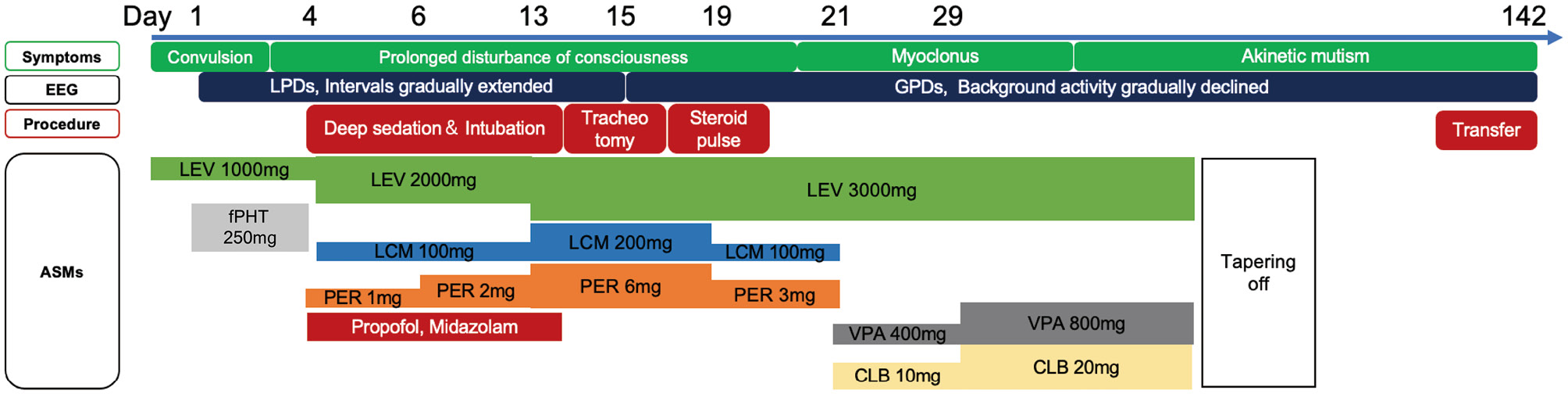
There were no abnormal findings on the cerebrospinal fluid (CSF) examination, and genetic panel testing for etiologic agents of central nervous system infections ruled out infectious meningitis or encephalitis. Head magnetic resonance imaging (MRI) showed decreased diffusion in the bilateral cerebral cortices (Fig. 3-A). On the 15th day of hospitalization, her EEG showed generalized periodic discharges (GPDs), which raised the suspicion of CJD (Fig. 1-B). Despite administering a steroid pulse as a diagnostic treatment for autoimmune encephalitis, there was no improvement. Testing for autoantibodies was also deemed negative (Table 1). Additionally, paraneoplastic limbic encephalitis was ruled out due to the absence of malignant findings in the trunk and negative results on antibody testing (Table 1). Around the 20th day of hospitalization, myoclonus subsequently developed in the left upper extremity and became generalized, and as a pyramidal sign, hyperreflexia of limb tendons predominantly in the left upper and lower extremities was also observed. Her head MRI revealed high intensity in the bilateral caudate nucleus and cerebral cortex in the diffusion-weighted image (Fig. 3-B), consistent with findings typically seen in CJD.3) Rapidly progressive cognitive impairment, characteristic neurologic findings such as myoclonus, pyramidal signs, and GPDs on EEG examination met the diagnostic criteria for "probable" sporadic CJD.4) Detection of specific 14-3-3 protein and high total tau protein levels in CSF, as well as abnormal prion protein detection via real-time quaking-induced conversion (RT-QUIC) method, also supported this diagnosis (Table 1). Myoclonus gradually disappeared, and the patient progressed to akinetic mutism approximately 1 month after hospitalization. MRI showed acute brain atrophy (Fig. 3-C), and EEG consistently demonstrated the presence of GPDs up to 128 days of hospitalization, while the intervals of discharges extended to approximately 3 seconds, concomitant with marked suppression of background activity (Fig. 1-C). On the 142nd day, the patient was transferred to a nursing care medical facility with a ventilator and remained in a state of akinetic mutism. Pathological examination will be considered at the time of death.
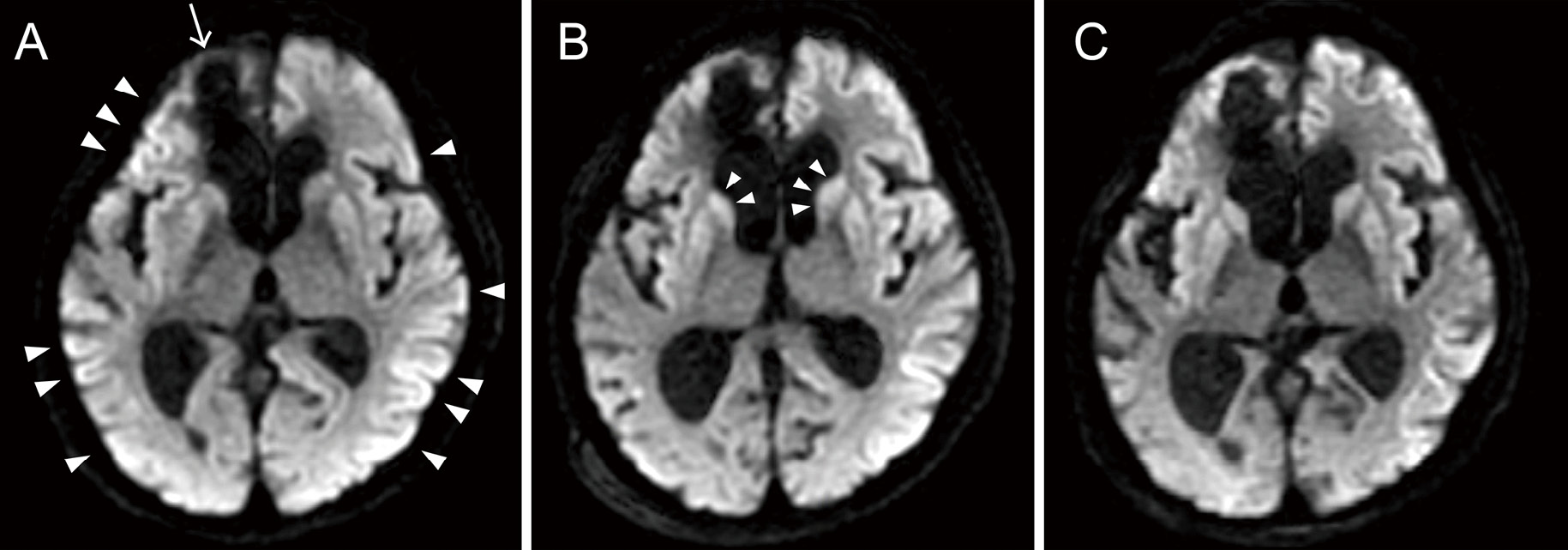
Table 1
Summary of test results for the patient with superrefractory status epilepticus
| Examination |
Results |
| Abbreviations: RT-QUIC, real-time quaking-induced conversion; AMPA-R Ab CBA, alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor antibody cell-based assay; AGNA-1, antiglial nuclear antibody-1; ANNA-1, antineuronal nuclear antibody; CASPR2, contactin-associated protein-like 2; CRMP-5-IgG, collapsin response-mediator protein-5; DPPX Ab IFA, dipeptidyl-peptidase-like protein-6 antibody immunofluorescence assay; GABA-B-R, gamma-aminobutyric acid receptor, type B; GAD65, antiglutamate decarboxylase antibody 65; GFAP, glial fibrillary acidic protein; IgLON5, neuronal cell adhesion molecule 5; LGI1, leucine-rich glioma inactivated 1; mGluR1, metabotropic glutamate receptor 1; NIF, neuronal intermediate filament; NMDA-R, N-methyl-D-aspartate receptor; PCA, Purkinje cytoplasmic antibody type 1; PCA-Tr, Purkinje cytoplasmic antibody type Tr |
| Cerebrospinal fluid examination |
| Appearance |
Clear |
| Cell count (cell/μL) |
<1 |
| Protein (mg/dL) |
49 |
| IgG index |
0.39 |
| Abnormal prion protein in cerebrospinal fluid and biomarker |
| total-Tau protein (pg/mL) |
Positive (>2200 pg/mL) |
| 14-3-3 protein |
Positive |
| RT-QUIC |
Positive |
| Encephalopathy, autoimmune/paraneoplastic evaluation |
| AMPA-R Ab CBA |
Negative |
| Amphiphysin Ab |
Negative |
| AGNA-1 |
Negative |
| ANNA-1 |
Negative |
| ANNA-2 |
Negative |
| ANNA-3 |
Negative |
| CASPR2 IgG CBA |
Negative |
| CRMP-5-IgG |
Negative |
| DPPX Ab IFA |
Negative |
| GABA-B-R Ab CBA |
Negative |
| GAD65 Ab Assay |
0 |
| GFAP IFA |
Negative |
| IgLON5 IFA |
Negative |
| LGI1-IgG CBA |
Negative |
| mGluR1 Ab IFA |
Negative |
| NIF IFA |
Negative |
| NMDA-R Ab CBA |
Negative |
| PCA-1 |
Negative |
| PCA-2 |
Negative |
| PCA-Tr |
Negative |
Discussion
The patient with focal epilepsy after SAH presented with disturbed consciousness and convulsion accompanied with LPDs on EEG and was initially diagnosed with SE of poststroke epilepsy. Although the convulsive seizures and EEG abnormalities improved after intravenous diazepam, the patient had prolonged disturbance of consciousness and recurrent LPDs on EEG examination, which later led to the diagnosis of sCJD. Indeed, epileptic seizures have been reported as an early manifestation of CJD, but because the patient had a history of poststroke epilepsy, other diagnoses were not considered until the patient progressed to a challenging course.
In this case, sCJD was strongly suspected due to the findings of progressive cognitive decline, GPDs on EEG examination, and abnormal MRI findings. In addition to the abovementioned symptoms, neurological findings such as myoclonus, pyramidal signs, akinetic mutism, and the specific 14-3-3 protein in the CSF and RT-QUIC result supported "probable" sCJD based on the diagnostic criteria proposed by Hermann P et al.4) Although this patient had a history of craniotomy, she had not undergone dura mater graft transplantation and was not considered to have iatrogenic CJD. Moreover, this patient did not show any psychiatric symptoms, did not have a disease duration of more than 6 months, and did not have pulvinar signs on MRI; therefore, the diagnosis of variant CJD was not considered.5) Based on these observations, the diagnosis was considered probable sCJD. Although autopsy results could provide additional insights, we have opted to present this case based on the available clinical and CSF findings. In Japan, the number of prion disease cases has been approximately 200 per year for the last 10 years, with sCJD accounting for 76% of the cases.6)
In sCJD, 0.4-3% of patients initially present with seizures, and 8-15% exhibit seizures during the entire course of the disease.7,8) Although sCJD is considered a diffuse brain disorder, the majority of convulsive seizures were reported to have developed as focal onset seizures.2) There have been previously documented cases of CJD presenting with lateralized neuropsychological symptoms.9,10) On the other hand, the patient, in this case, was reported to have poor medication adherence during the course of the disease, convulsive seizures, and LPDs on EEG examination with maximum amplitude in the right frontal region, which is anatomically concurrent with the location of brain damage after SAH. Therefore, it was difficult to distinguish whether the initial convulsive seizures were one of the symptoms of sCJD or SE of poststroke epilepsy. Considering the prevalence of CJD, it would not have been practical to suspect CJD in the early stages of this case. With regard to the MRI findings, it is recognized that epileptic seizures can be accompanied by decreased diffusion in the cortex or striatum.11) In cases in which seizures are present at the time of MRI imaging, it is difficult to distinguish whether the MRI findings are changes due to seizures or CJD. In this present case, the MRI findings led us to suspect CJD as there were no obvious seizures immediately before or during MRI imaging.
The patient exhibited LPDs on EEG examination in the early stage and later evolved to diffuse periodic sharp wave complex (PSWC), which was generally the typical EEG findings in CJD. PSWCs are characterized by the presence of simple sharp waves (including biphasic and triphasic waves) with a predominant bilateral voltage distribution and a front-precentral midline maximum.8) This EEG finding is also commonly referred to as GPDs or periodic synchronous discharges. Previous literature has also reported that LPDs occur in the early stages of CJD prior to GPDs.12-17) LPDs are generally recognized as occurring in cases of acute or subacute unilateral cerebral lesions, such as strokes. In contrast to CJD-associated PSWCs, LPDs are typically transient EEG findings and resolve within 2 weeks, characterized by a decrease in amplitude and periodicity over the course of the disease.18) On the other hand, it has been widely reported that lateralized PSWCs initially observed in CJD patients consistently evolve into bilateral PSWCs.8) Au et al. have also reported three cases manifesting LPDs on EEG examination in the early stages of CJD, and the duration of LPDs ranged from 6 to 21 days in those cases. Considering that LPDs in most etiologies are transient EEG abnormalities, Au et al. recommended suspecting CJD in cases with persisting LPDs.13) As CJD is known to be rapidly degenerative and uniformly fatal, early detection of clinical findings is crucial to prevent further transmission.
The patient was in convulsive SE at admission; therefore, the subsequent prolonged comatose state with persistent LPDs on EEG examination was also thought to be due to SE. Clinical manifestation and EEG abnormalities did not improve with deep sedation, a third-line ASM; therefore, superrefractory SE was considered. It was thought that the primary cause of SE is remote stroke or low ASM level,19,20) and the etiology of "refractory" SE appears to be similar overall to that of nonrefractory SE.21) On the other hand, the etiology of "superrefractory" SE may be different from that of SE and refractory SE.22) Several studies have suggested that encephalitis is a frequent cause of superrefractory SE.23-25) Therefore, if clinical manifestations and EEG abnormalities do not improve even after third-line ASM, it is necessary to consider a diagnosis other than SE due to a major cause (in this case, poststroke epilepsy). Although this case did not meet the criteria for new-onset refractory status epilepticus (NORSE)26) as it was not a newly onset epilepsy, various differential diagnoses, including encephalitis, central nervous system infection, and CJD, should be considered in a similar approach taken with NORSE.
In cases of superrefractory SE, it is essential to investigate the underlying causes of acute symptomatic seizures, including CJD or various types of encephalitis. Furthermore, in the early stages of CJD, LPDs may be present on EEG examination, and lateralized neurological symptoms such as focal onset seizures may be observed. Therefore, it is crucial to consider CJD as a differential diagnosis in such cases.
Ethical Statement
Informed consent was obtained from the patient's family.
Funding
This work was not supported by any funding agency.
Conflicts of Interest Disclosure
The authors declare no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- 1) Abrahantes JC, Aerts M, Van Everbroeck B, et al.: Classification of sporadic Creutzfeldt-Jakob disease based on clinical and neuropathological characteristics. Eur J Epidemiol 22: 457-465, 2007
- 2) Mbizvo GK, Ziso B, Larner AJ: Epilepsy and prion diseases: A narrative review. Epilepsy Behav 115: 107630, 2021
- 3) Eisenmenger L, Porter MC, Carswell CJ, et al.: Evolution of diffusion-weighted magnetic resonance imaging signal abnormality in sporadic Creutzfeldt-Jakob disease, with histopathological correlation. JAMA Neurol 73: 76-84, 2016
- 4) Hermann P, Appleby B, Brandel JP, et al.: Biomarkers and diagnostic guidelines for sporadic Creutzfeldt-Jakob disease. Lancet Neurol 20: 235-246, 2021
- 5) European Centre for Disease Prevention and Control: Variant Creutzfeldt-Jakob disease, EU case definition. https://www.ecdc.europa.eu/en/infectious-disease-topics/z-disease-list/variant-creutzfeldt-jakob-disease/eu-case-definition (Accessed Aug 25 2021)
- 6) Masahito Y: Researches on rare and intractable diseases health, labour and welfare policy research grants The Ministry of Health, Labour and Welfare, Japan The annual report of the Research Committee on Surveillance and Infection Control of Prion Disease in FY2021. 2022
- 7) Brown P, Cathala F, Castaigne P, Gajdusek DC: Creutzfeldt-Jakob disease: Clinical analysis of a consecutive series of 230 neuropathologically verified cases. Ann Neurol 20: 597-602, 1986
- 8) Wieser HG, Schindler K, Zumsteg D: EEG in Creutzfeldt-Jakob disease. Clin Neurophysiol 117: 935-951, 2006
- 9) Cambier DM, Kantarci K, Worrell GA, Westmoreland BF, Aksamit AJ: Lateralized and focal clinical, EEG, and FLAIR MRI abnormalities in Creutzfeldt-Jakob disease. Clin Neurophysiol 114: 1724-1728, 2003
- 10) Mourand I, Grandiere-Perez L, Joyes B, Legout A: Lateralized clinical and diffusion-weighted MRI abnormalities in a probable case of sporadic Creutzfeldt-Jakob disease. Acta Neurol Belg 110: 193-195, 2010
- 11) Fujita K: Neuroimaging in the differential diagnosis of prion disease. Rinsho Shinkeigaku 23: 1249-1251, 2013
- 12) Fushimi M, Sato K, Shimizu T, Hadeishi H: PLEDs in Creutzfeldt-Jakob disease following a cadaveric dural graft. Clin Neurophysiol 113: 1030-1035, 2002
- 13) Au WJ, Gabor AJ, Vijayan N, Markand ON: Periodic lateralized epileptiform complexes (PLEDs) in Creutzfeldt-Jakob disease. Neurology 30: 611-617, 1980
- 14) Eggertson DE, Pillay N: Creutzfeldt-Jakob disease: Correlation of focal electroencephalographic abnormalities and clinical signs. Can J Neurol Sci 13: 120-124, 1986
- 15) Primavera A, Tabaton M, Leonardi A: Periodic lateralized discharges in Creutzfeldt-Jakob disease: Serial electroencephalographic studies. Rev Electroencephalogr Neurophysiol Clin 13: 379-382, 1984
- 16) Heye N, Cervós-Navarro J: Focal involvement and lateralization in Creutzfeldt-Jakob disease: Correlation of clinical, electroencephalographic, and neuropathological findings. Eur Neurol 32: 289-292, 1992
- 17) Heye N, Henkes H, Hansen ML, Gosztonyi G: Focal-unilateral accentuation of changes observed in the early stage of Creutzfeldt-Jakob disease. J Neurol Sci 95: 105-110, 1990
- 18) Westmoreland BF, Klass DW, Sharbrough FW: Chronic periodic lateralized epileptiform discharges. Arch Neurol 43: 494-496, 1986
- 19) DeLorenzo RJ, Hauser WA, Towne AR, et al.: A prospective, population-based epidemiologic study of status epilepticus in Richmond, Virginia. Neurology 46: 1029-1035, 1996
- 20) Knake S, Rosenow F, Vescovi M, et al.: Incidence of status epilepticus in adults in Germany: A prospective, population-based study. Epilepsia 42: 714-718, 2001
- 21) Nelson BSE, Varelas PN: Status epilepticus, refractory status epilepticus, and super-refractory status epilepticus. CONTIN Lifelong Learn Neurol 24: 1683-1707, 2018
- 22) Hocker S, Tatum WO, LaRoche S, Freeman WD: Refractory and super-refractory status epilepticus: An update. Curr Neurol Neurosci Rep 14: 1-9, 2014
- 23) Tian L, Li Y, Xue X, et al.: Super-refractory status epilepticus in West China. Acta Neurol Scand 132: 1-6, 2015
- 24) Jayalakshmi S, Ruikar D, Vooturi S, et al.: Determinants and predictors of outcome in super refractory status epilepticus-A developing country perspective. Epilepsy Res 108: 1609-1617, 2014
- 25) Holtkamp M, Othman J, Buchheim K, Masuhr F, Schielke E, Meierkord H: A "malignant" variant of status epilepticus. Arch Neurol 62: 1428-1431, 2005
- 26) Costello DJ, Kilbride RD, Cole AJ: Cryptogenic new onset refractory status epilepticus (NORSE) in adults-infectious or not? J Neurol Sci 277: 26-31, 2009