Reviews
Millisecond Ca2+ dynamics activate multiple protein cascades for synaptic vesicle control
2017 年 93 巻 10 号 p. 802-820
詳細
2017 年 93 巻 10 号 p. 802-820
For reliable transmission at chemical synapses, neurotransmitters must be released dynamically in response to neuronal activity in the form of action potentials. Stable synaptic transmission is dependent on the efficacy of transmitter release and the rate of resupplying synaptic vesicles to their release sites. Accurate regulation is conferred by proteins sensing Ca2+ entering through voltage-gated Ca2+ channels opened by an action potential. Presynaptic Ca2+ concentration changes are dynamic functions in space and time, with wide fluctuations associated with different rates of neuronal activity. Thus, regulation of transmitter release includes reactions involving multiple Ca2+-dependent proteins, each operating over a specific time window. Classically, studies of presynaptic proteins function favored large invertebrate presynaptic terminals. I have established a useful mammalian synapse model based on sympathetic neurons in culture. This review summarizes the use of this model synapse to study the roles of presynaptic proteins in neuronal activity for the control of transmitter release efficacy and synaptic vesicle recycling.
Neuronal activity, in the form of action potentials (APs) conducted to the presynaptic terminal, triggers the release of neurotransmitters into the synaptic cleft to activate postsynaptic receptors, resulting in chemical synaptic transmission. Since Sir Bernard Katz proposed the “vesicular theory”1) and reported the role of Ca2+ ions in mediating neurotransmitter release,2) accumulated evidence has established that presynaptic APs opens Ca2+ channels, and Ca2+ entry initiates the release of neurotransmitters. Neurotransmitter release occurs at specialized sites called the active zone (AZ), enriched with many proteins, including Ca2+ channels. In the AZ, synaptic vesicles (SVs) filled with neurotransmitters dock at the plasma membrane and are ready for exocytosis upon Ca2+ influx.3),4) However, how Ca2+ ions trigger neurotransmitter release had been unknown till molecular biologists started identifying presynaptic proteins in the early 1990s.
To understand the molecular control of SV dynamics, it is necessary to alter the molecular constituents of presynaptic terminals, while simultaneously measuring the functional consequences of such molecular perturbations. However, it is challenging to do such functional studies, because (1) the reactions to be studied often occur over time scales of milliseconds; and (2) presynaptic terminals are small and difficult to study. The solution to problem (1) is to employ electrophysiological measurements, which Katz and others have shown to have the requisite time resolution. However, because of problem (2), such functional studies are most readily applied to large presynaptic terminals,5),6) which were rare for the case of mammalian synapses in the early 1990s.
Presynaptic function has classically been studied by electrophysiological measurements using invertebrate synapses, such as the squid stellate ganglion giant synapse5) and Aplysia ganglionic synapses.6),7) The large size of their presynaptic elements make it possible to microinject agents and determine their effects on synaptic transmission. I had been trained in electrophysiological measurements of synaptic transmission in cat, rabbit, and rat sympathetic ganglia by Haruo Kobayashi in Japan as a research fellow and by Benjamin Libet in the US as a postdoctoral fellow, examining postsynaptic plasticity (Benjamin Libet, in The history of neuroscience in autobiography, volume 1, 414–453, Ed. Larv R. Squire Washington, DC, Society for Neuroscience, 1996). The influence of the work of Peter Baker in the U.K. led to a shift in my research interests towards the mechanism of neurotransmitter release.8) He was a pioneer in the study of Ca2+ homeostasis, and to understand mechanisms of secretory responses, he had started to apply botulinum neurotoxin. Sadly, he passed away at 47 years old in 1987. Between 1988 and 1990, I joined the laboratory of Ladislav Tauc in France. During this time, in collaboration with toxicologists, I studied the molecular mechanism of transmitter release blockade after tetanus and botulinum neurotoxin mRNA expression in Aplysia synapses.7) This was the first study in the world to show the effect of newly synthesized proteins in living neurons, with mRNA injection inducing a block of synaptic transmission during electrophysiological recordings. Such was the excitement at obtaining these results, Professor Tauc opened a bottle of champagne around the rig where we had been observing the decrease in transmitter release. Since then, I have often visited Heiner Niemann’s laboratory in Tubingen, Germany, to examine mutant mRNAs.
My experience in studying neurotoxin action in Aplysia synapses was crucial in allowing me to establish a useful mammalian synaptic system after my return to Japan. The superior cervical ganglion (SCG) neurons of juvenile rats, which form a well-characterized cholinergic synapse in long-term cultures,9),10) are an ideal cell model11) with which one can investigate multiple protein functions in transmitter release processes (Fig. 1).9),12) The SCG neuron has a large cell body and nucleus that allows for the manipulation of gene expression and protein function in mature neurons via acute microinjection of cDNA, small interfering RNA (siRNA), dominant-negative transgenes, peptides, antibodies, and/or metabolites,9),13)–16) an approach not technically feasible for cultured neurons from the central nervous system. In addition, synaptic activity and short-term plasticity, because they relate to the size and replenishment of functional SVs pools, can be accurately monitored by recording excitatory postsynaptic potentials (EPSPs) evoked by APs in presynaptic neurons.

An experimental system to study presynaptic proteins function in a fast synapse formed between superior cervical ganglion (SCG) neurons in culture. A, Phase contrast image of SCG neurons cultured for 5 weeks. Cells were prepared from 7-day-postnatal rats. B, Schematic drawing of electrophysiological recording and agent injection. Action potential (AP) generated by current injection through a sharp electrode releases neurotransmitters from presynaptic terminals, and the excitatory postsynaptic potential (EPSP) is recorded by another sharp electrode. Various agents can be dialyzed into the large cell body, along with color dye for confirming the injection, and rapidly diffuse to presynaptic terminals. Effects of the injected agent are monitored by changes in the EPSP waveform. C, Change in the EPSP waveform (left, one representative experiment) and reduction in EPSP amplitude (right, average of 5–7 experiments) by an N-type Ca2+ channel synprint peptide that disrupts the interaction with the SNARE proteins complex (see Fig. 2). 65 µM (left traces; △), 130 µM synprint peptide (▲) (see Fig. 2B), or as a control, 140 µM peptide similar site of L-type channels (◇) was injected at time = 0 for 2–3 min during EPSP recording every 20 s.
Using this approach, and collaborating with molecular biologists in the US, Europe, and Japan, I have elucidated the functions of numerous presynaptic proteins.11),13)–27) An example of my early studies on the microinjection of peptides prepared for a protein–protein interaction site is described in the next section 2. I hypothesized that changes in presynaptic Ca2+ concentration after AP firing activates multiple Ca2+-sensing proteins to control SVs dynamics. Thus, the goal of my work was to establish the molecular basis for Ca2+ signaling within the presynaptic terminal. Section 3 focuses on my studies using a combination of genetic manipulations of key proteins to affect neural activity-dependent regulation of transmitter release. My studies have revealed that regulation of neurotransmitter release requires fine control of ultrafast protein reactions that are activated by a diverse range of Ca2+-activated effectors.12) In section 4, a different approach to the study molecular mechanisms in neurotransmitter release is discussed.
The “vesicular theory” proposed by Katz suggested synchronization of transmitter release from SVs,1) mediated by Ca2+ ions.2) Further studies on the dependence on Ca2+ ions suggested the presence of Ca2+ sensors. Forty years after the proposal of the “vesicular theory”, a SV protein family called synaptotagmins was identified as Ca2+ sensors28) and their functions were demonstrated in the early 1990s.29) Since then, the molecular mechanisms of synaptotagmin action have been studied in numerous laboratories, including mine in collaboration with Katsuhiko Mikoshiba at the University of Tokyo, Japan.30) We now know that synaptotagmins are a low-affinity type of Ca2+ binding protein, and they serve as Ca2+ sensors for fast neurotransmitter release. Binding of synaptotagmin isoforms 1 and 2 to the mouth of Ca2+ channels ensures the fast synchronization of SVs exocytosis, in cooperation with a protein in SVs and two proteins in the plasma membrane, called SNARE (soluble N-ethylmaleimide-sensitive factor attachment protein receptor) proteins, for their fusion.30)–34)
The presynaptic Ca2+ channel family encodes the pore-forming subunit (α1 subunit, Fig. 2A).35) William Catterall and colleagues in the US found that the N-type Ca2+ channel interacts with the SNARE protein complex at an intracellular loop of the pore-forming subunit, named the ‘synprint’ (synaptic protein interaction) site (Fig. 2B).36) This interaction is Ca2+ dependent, with maximal binding at 20 µM Ca2+, which is an optimal Ca2+ concentration for SVs exocytosis, and reduced binding at lower or higher Ca2+ concentrations, suggesting that the precise concentration of presynaptic Ca2+ regulates the association and dissociation of SNARE proteins with presynaptic Ca2+ channels.
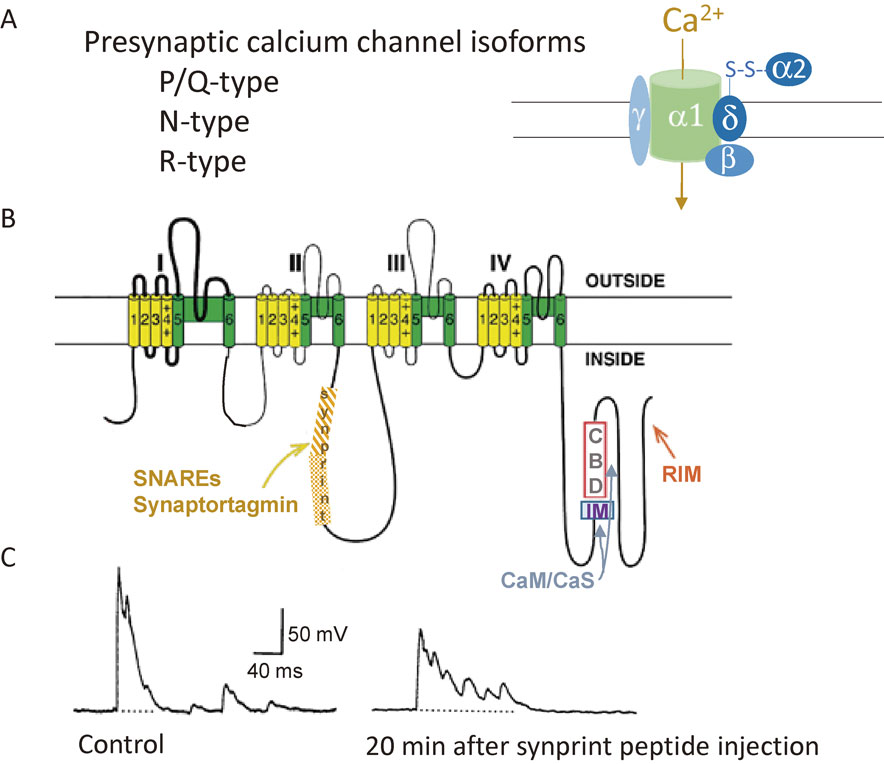
Ca2+ channel structure, organization, and interaction with regulatory proteins. A, Overview of presynaptic Ca2+ channel isoforms. Representation of subunit composition of Ca2+ channels and auxiliary subunits. B, Assumed membrane topology of Ca2+ channel α1 subunit composition. The α1 subunit consists of four homologous domains (I–IV), each consisting of six transmembrane segments (S1–S6). Cylinders represent predicted helices. S1–S4, shown in yellow, represents the voltage-sensing module. S5–S6, shown in green, represents the pore-forming unit. The large intracellular loops linking the different domains of the α1 subunit serve as sites of interaction with channel activity regulatory proteins, such as SNAREs and synaptotagmin, calmodulin (CaM), Ca2+ sensor proteins (CaSs) and RIM, an active zone protein. Adapted from Catterall and Few, 2008. C, Injection of a peptide shown with shaded region in the cytoplasmic II–III loop (B), named the ‘synprint’ site, reduced the fast synchronous neurotransmitter release but increased asynchronous release in higher 5 mM Ca2+ extracellular solution. Adapted from Mochida et al., 1996.
To examine the physiological significance of the interaction, Catterall and colleagues needed an ideal synapse to introduce peptides derived from the ‘synprint’ site, which competitively inhibited interactions between Ca2+ channels and SNARE proteins in vitro, into the presynaptic terminal. Catterall was informed about “my model synapse” by Masami Takahashi, who found the interaction, and sent the ‘synprint’ peptides to my laboratory. I injected the peptides into presynaptic SCG neurons, monitoring synaptic transmission by recording EPSPs (Fig. 1B). ‘Synprint’ peptides significantly reduced synaptic transmission (Figs. 1C, 2C) by uncoupling the endogenous interaction of the Ca2+ channel–SNARE protein complex at nerve terminals.22) Surprisingly, late asynchronous release increased, whereas rapid and synchronous synaptic transmission was selectively inhibited following the injection (Fig. 2C). Thus, we proposed that coupling of Ca2+ channels to SVs via SNARE proteins complex is essential for synchronization of SV exocytosis and efficient synaptic transmission.
Later, a requirement for close coupling of Ca2+ channels to SVs for the efficient release of neurotransmitters also emerged from studies using giant squid synapses37) and the calyx of Held.38),39) The high efficiency of P/Q-type Ca2+ currents in initiating neurotransmitter release is correlated with the close localization of docked vesicles near the Ca2+ channels.38),40)
The synchronization of SV exocytosis is regulated by the precise concentration of presynaptic Ca2+. This fact led to a fascinating idea that the dynamic range of Ca2+ concentration after AP firing activates multiple Ca2+ sensors as switches of SV control for the next round of transmitter release.41) Several Ca2+-binding proteins, which sense residual Ca2+ that diffuses away from Ca2+ channels after the firing of APs, may act as potential effectors for these reactions.42) For example, considerable evidence supports a role for calmodulin (CaM), a higher-affinity Ca2+-binding protein, in the modulation of release efficacy by binding to SNARE proteins,27),43) regulation of SV recycling,44),45) and mobilization of SVs, which was the focus for my first study on presynaptic protein after my return to Tokyo.11) The following sections 3.1–3.3 summarize the regulation of key proteins by Ca2+ dynamics accompanying Ca2+ entry, such as Ca2+ channels that control the efficacy of transmitter release from SVs (section 3.1), AZ proteins that dock SVs close to Ca2+ channels (section 3.2), and motor proteins that mobilize SVs to the docking site (section 3.3).
3.1 Regulation of neurotransmitter release efficacy — Modulation of Ca2+ channels —.After briefly describing the background of this study, section 3.1 summarizes a decade of my studies on the regulation of neurotransmitter release by Ca2+-dependent Ca2+ channel modulation in collaboration with William Catterall. I expressed brain-derived P/Q type-Ca2+ channels in presynaptic SCG neurons by microinjection of cDNA (Fig. 3), prepared in the Catterall laboratory,16),22),46),47) and examined the mediation of synaptic transmission using electrophysiological measurements (Fig. 4). Surprisingly, a single AP, within tens of milliseconds, activated CaM and certain Ca2+ sensor proteins with different calcium affinities and binding speeds, which modulated Ca2+ channel opening, thereby resulting in time to time fine tuning of transmitter release efficacy (Fig. 4).13),17)
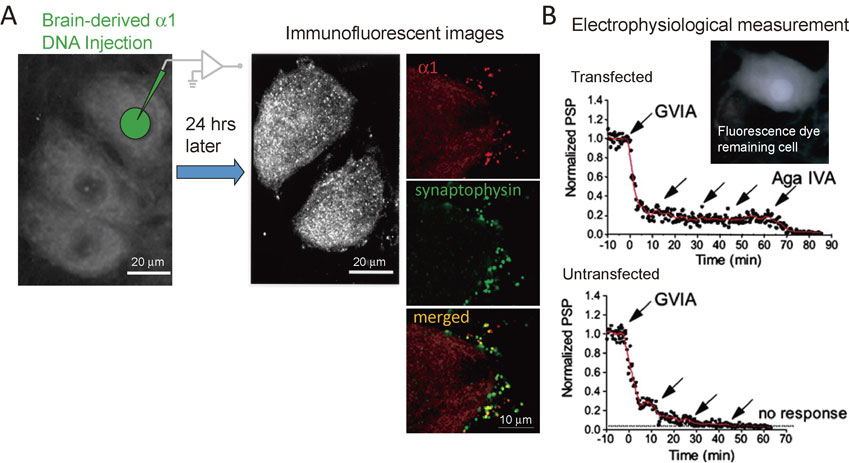
Combination of genetic manipulation of a key protein and electrophysiological measurements of synaptic transmission. A, Expression of brain-derived Ca2+ channels. Imunofluorescent images of the α12.1 subunit. Left, diagram of cDNA injection into a nucleus. cDNA was dissolved in the suction pipette solution (150 mM potassium acetate/5 mM MgATP/10 mM Hepes, pH 7.35). The cDNAs were introduced into the nucleus of superior cervical ganglion (SCG) neurons by diffusion from a glass pipette (40- to 60-MΩ tip resistance) with hand pressure applied to a syringe connected to the injection pipette. The concentration of cDNAs in the injection pipette was 0.275 mg/ml. Dextran fluorescein (10 kDa) was introduced in the pipette solution to monitor cDNA entry into nuclei. Right, 24 hours later, an antibody against the α12.1 subunit showed newly expressed α1 subunit in the cell body (white dots) and the presynaptic terminals (α1, red dots) colocalizing with a synaptic vesicle protein, synaptophysin. B, Excitatory postsynaptic potential (EPSP) reduction with bath application of selective Ca2+ channel blockers (arrows). An action potential was generated in the fluorescence dye remaining cell body and EPSP was recorded from an adjacent uninjected neuron. An arrow indicates drop application of 2.5 µM GVIA for N-type channels, or 0.25 µM Aga IVA for P/Q-type channels block. In the transfected synapse, EPSP remaining in the presence of GVIA disappeared with Aga IVA (upper). In contrast, EPSP disappeared completely with GVIA in an untransfected synapse. These results suggest exogenous α1 subunit forms functional Ca2+ channels. Adapted from Mochida et al., 2003.

Action potential (AP) firing modulates synaptic transmission mediated by P/Q-type Ca2+ channels. P/Q-type Ca2+ channels, wild-type (WT), alanine mutation at the IQ-like motif (IM) motif, and deletion of the calmodulin (CaM) binding domain (ΔCBD) were expressed in presynaptic neurons and the synaptic transmission was examined in the presence of a blocker for native N-type Ca2+ channels. A, Paired-AP alters the second excitatory postsynaptic potential (EPSP). Left, the second AP with a 80 ms interval (upper trace) induces a larger EPSP (lower trace), i.e., synaptic facilitation. Right, timing of the second AP-induced depression and facilitation of synaptic transmission. Depression was prevented by ΔCBD, while facilitation was prevented by the IM-AA mutation of the pore-forming subunit. B, Biphasic synaptic transmission during 1-s AP bursts at 30 Hz changed to synaptic depression by IM-AA or synaptic facilitation by ΔCBD mutation. Adapted from Mochida et al., 2008. C, Model for Ca2+/CaM-dependent inactivation and facilitation of P/Q-type of Ca2+ channels and neurotransmitter release.
During our long collaboration, the functional study system with cultured SCG neuron synapses (Fig. 1B) was also established in the Catterall laboratory, in addition to the laboratories of Zuhang Sheng in NIH and Michael Seager in France, who had studied in the Catterall laboratory.
3.1.1 Ca2+ sensors regulate Ca2+ channel activity (background to this study).The Ca2+ channel proteins consists of a pore-forming α1 subunit associated with β, α2-δ, and possibly γ subunits (Fig. 2A).48) Catterall and colleagues found that repetitively generated Ca2+ currents increased and then decreased due to the channel modulation by Ca2+ elevation.49) Ca2+ elevation at the mouth of P/Q-type Ca2+ channels regulated the channel activity by binding to CaM35),49)–52) and to the related neuron-specific Ca2+-binding proteins, Ca2+-binding protein-1 (CaBP1) and visinin-like protein-2 (VILIP-2).53)–55) CaM and the Ca2+-binding proteins interact with a bipartite regulatory site of the α1 subunit52) called the IQ-like motif (IM) and the nearby CaM-binding domain (CBD; Fig. 2B). Alanine substitutions in the IM (IM-AA) blocked Ca2+-dependent facilitation of Ca2+ channels,50),52) whereas Ca2+-dependent inactivation was blocked by channels lacking the CBD (ΔCBD).49),51),52),54),55)
3.1.2 Combination of genetic manipulation of Ca2+ channels and electrophysiological measurements of synaptic transmission.Analyzing P/Q-type channel properties is difficult, because at most presynaptic terminals, Ca2+ channels are expressed diversely, comprising three different types of channels, N, P/Q, and R. In contrast, synaptic transmission of SCG neurons is mediated by N-type channels.21) Thus, in the presence of N-type channel blockers, SCG neuron synapses expressing brain-derived P/Q-type channels in the presynaptic neuron are a useful means of elucidating the role of CaM-mediated modulation of P/Q-type Ca2+ channels in synaptic transmission.
A cDNA encoding the rat brain Ca2+-channel subunit α12.1, which forms the P/Q-type Ca2+ channel, was subcloned into a eucaryotic expression vector.16) cDNA (3–6 mg/ml) was shipped on dry ice from Seattle to Tokyo and stored at −80 °C until use. The cDNAs were introduced into the nucleus of SCG neurons by diffusion from a glass pipette (40- to 60-MΩ tip resistance with 150 mM potassium acetate/5 mM MgATP/10 mM Hepes, pH 7.35) with hand pressure applied to a syringe connected to the injection pipette. The concentration of cDNAs in the injection pipette was 0.275 mg/ml. Dextran fluorescein (10 kDa, Molecular Probes) was introduced in the pipette solution to monitor cDNA entry into nuclei (Fig. 3A). Expression of the cDNA was confirmed by immunohistochemistry (Fig. 3A).
For electrophysiologyical studies of synaptic transmission, 1 day after the injection of cDNAs, the injected nucleus and cell body showing fluorescent dye was used as a presynaptic neuron to generate APs with current injection. The EPSP was recorded from an adjacent untransfected neuron. Expression of a functional Ca2+ channel was confirmed by bath application of a selective Ca2+ channel blocker, GVIA for N-type channel blocking or Aga IVA for P/Q-type channel blocking (Fig. 3B). EPSP disappeared with GVIA application in untransfected synapses. However, in cDNA-transfected synapses, 20–30% of the remaining EPSP amplitude in the presence of GVIA disappeared with Aga IVA application, suggesting that P/Q-type channels were functionally expressed together with other endogenous subunits.16)
3.1.3 Ca2+/CaM-Ca2+ channel interaction mediates synaptic depression and facilitation.To elucidate the role of CaM-mediated modulation of P/Q-type Ca2+ channels in synaptic transmission, in the presence of an N-type channels blocker, SCG neuron synapses expressing brain-derived P/Q-type channels and mutants in the presynaptic neuron were used for electrophysiological analysis.
After AP firing, Ca2+ elevation induces the bidirectional modulation of Ca2+ channel activity via binding of Ca2+-CaM (Fig. 4A, C).13) CaM possesses four EF-hand Ca2+-binding motifs organized in two lobes. The N-lobe of CaM senses rapid and higher increases in presynaptic Ca2+ concentration56) to initiate Ca2+ channel inhibition, resulting in synaptic depression. In contrast, synaptic facilitation during enhancement of channel activity is mediated by the C-lobe of CaM sensing a lower Ca2+ concentration.56) These temporal dynamics can be observed by monitoring EPSPs evoked by pairs of APs, and depending on the interval between APs (paired pulse interval [PPI]), either synaptic depression or facilitation is observed (Fig. 4A). Synaptic depression (i.e., 1st EPSP > 2nd EPSP) takes place when the PPI is <50 ms, whereas synaptic facilitation (i.e., 1st EPSP < 2nd EPSP) occurs for PPI between 50 and 100 ms (Fig. 4A, C). Thus, time-dependent regulation of synaptic transmission due to bidirectional modulation of Ca2+ channel activity is induced by binding of CaM, which senses the local Ca2+ concentration at the mouth of Ca2+ channels.
In vivo, neural information is encoded in bursts of APs. During such bursts, the efficacy of neurotransmitter release gradually decreases (Fig. 4B, WT). This ‘synaptic depression’ is thought to be due to the depletion of SVs in the presynaptic terminal.2) However, I found that the ‘synaptic depression’ was not due to SV depletion, but instead was induced by Ca2+ channel regulation by Ca2+-CaM. Deletion of the CBD reversed the depression and instead resulted in facilitation (Fig. 4B, ΔCBD), i.e., following AP bursts, CaM binding to the CBD down-regulated the release efficacy, resulting in synaptic depression. In contrast, a mutation in the IM (Fig. 4B, IM-AA) potentiated the synaptic depression, i.e., CaM binding to the IM up-regulated the release efficacy, resulting in synaptic facilitation. The facilitation with CBD deletion lasted throughout a 1 s AP burst (Fig. 4B), indicating sufficient storage of SVs for a short burst of APs.
This regulation of P/Q-type Ca2+ channels has recently been confirmed in short-term plasticity in native hippocampal synapses by Catterall and colleagues.57) Of note, however, endogenous N-type Ca2+ channels mediating transmitter release from SCG neurons showed Ca2+-CaM-dependent synaptic depression due to inhibition of channel activity but not Ca2+-CaM-dependent synaptic facilitation.20)
3.1.4 CaBP1 and VILIP-2-binding to the same sites of Ca2+ channels as CaM-binding mediate bidirectional synaptic modulation.CaM-like Ca2+ sensor proteins (CaSs) are expressed in neurons. Catterall and colleagues found that, among CaSs expressed in the brain,58),59) CaBP1 binds to the CBD,55) whereas VILIP-2 binds to the CBD and IM of P/Q-type Ca2+ channels.54)
To examine the physiological roles of the interactions of CaBP1 or VILIP-2 with P/Q-type Ca2+ channels, each CaS was expressed together with P/Q-type Ca2+ channels in presynaptic SCG neurons, and synaptic transmission was monitored in the presence of a native Ca2+ channel blocker.17) In transfected SCG neurons, CaBP1 and VILIP-2 altered synaptic transmission mediated by P/Q-type Ca2+ channels. Whereas CaBP1 enhanced synaptic depression,17) VILIP-2 facilitated synaptic transmission.17) CaS-dependent synaptic modulation was absent when Ca2+ channels had an impaired IM motif (AA mutation) or CBD deletion.17)
Expression levels of CaBP1, VILIP-2, and CaM in the brain differ locally,58),59) suggesting that regulation of presynaptic Ca2+ channels by CaSs and CaM may play a critical role in determining the diversity of short-term synaptic plasticity at each synapse expressing high levels of CaBP1, VILIP-2, or CaM.
3.1.5 Regulation of transmitter release efficacy by multiple Ca2+ sensors binding to Ca2+ channels.As described above, electrophysiological analyses revealed surprisingly fast reactions of Ca2+ effectors. An AP, within tens of milliseconds, activates CaM, CaBP1, and VILIP-2 to modulate Ca2+ channel activity, resulting in immediate fine tuning of neurotransmitter release efficacy.13),17) Here, I would like to discuss why multiple Ca2+-binding proteins regulate the activity of Ca2+ channels at the same sites as CaM. The opening of Ca2+ channels creates a steep gradient of increased Ca2+ concentration at the SV release site, where each Ca2+ sensor has different affinity and binding speed to Ca2+:56) CaM (5–10 µM) > CaBP1 (2.5 µM) > VILIP-2 (∼1 µM),60) suggesting that their affinity and kinetics of Ca2+ binding determine the timing of Ca2+ channel modulation, resulting in temporal regulation of transmitter release efficacy. Actually, after AP firing, the effective time for the regulation of transmitter release efficacy by CaM and CaSs is different: CaM action occurs shortly after Ca2+ entry and lasts 100 ms (Fig. 4A), whereas the actions of CaBP1 or VILIP-2 last longer, 150 or 250 ms.17) Expression levels of CaBP1, VILIP-2, and CaM in the brain differ locally.58),59) The divergent actions of CaM and CaSs on P/Q-type Ca2+ channels, which are dependent on the rate of Ca2+ reduction, may fine-tune the regulatory properties of presynaptic Ca2+ channels, allowing a greater range of input–output relationships and short-term plasticity at different synapses.61)
3.2 Regulation of SV docking close to Ca2+ channels — Phosphorylation of an AZ protein —.Residual Ca2+ after AP firing accumulates and activates protein kinases that modulate the property of SNARE proteins62) and Ca2+ sensors,63) resulting in the modulation of transmitter release probability. In collaboration with Masamichi Igarashi at Niigata University, Japan, and Yoshimi Takai at Kobe University, Japan, I also showed in 2002 and 2005 that Ca2+/ATP-dependent CaMKII binding to a SNARE protein, and phosphorylation of a protein that binds to the SNARE complex, upregulate transmitter release efficacy.14),64) For rapid remodeling of synaptic efficacy, in addition to the modulation of proteins related to SV exocytosis, phosphorylation of protein(s) forming the platform of SVs for exocytosis is ideal. In this section 3.2, after a brief introduction to AZ proteins (section 3.2.1) and phosphorylation (section 3.2.2), I summarize more than a decade of studies on AZ proteins in the regulation of AZ architecture and SV recruitment, performed in collaboration with Toshihisa Ohtsuka, Yamanashi University, Japan.18),65),66)
3.2.1 SV docking and Ca2+ channel recruitment by AZ proteins (background to this study 1).The AZ is a slightly electron dense region beneath the presynaptic plasma membrane where SVs dock, fuse, and release their neurotransmitter contents into the synaptic cleft (Fig. 5A). The location of Ca2+ channels and the docking of SVs to the release site are regulated by an AZ protein, RIM (Rab3-interacting molecule) (Fig. 5B).67) RIM interacts with the C-terminal sequences of Ca2+ channels (Fig. 2B)68),69) and other AZ proteins (Fig. 5B). A molecular complex consisting of RIM and the C-terminal tails of the Ca2+ channels determine the recruitment of Ca2+ channels to the AZ.
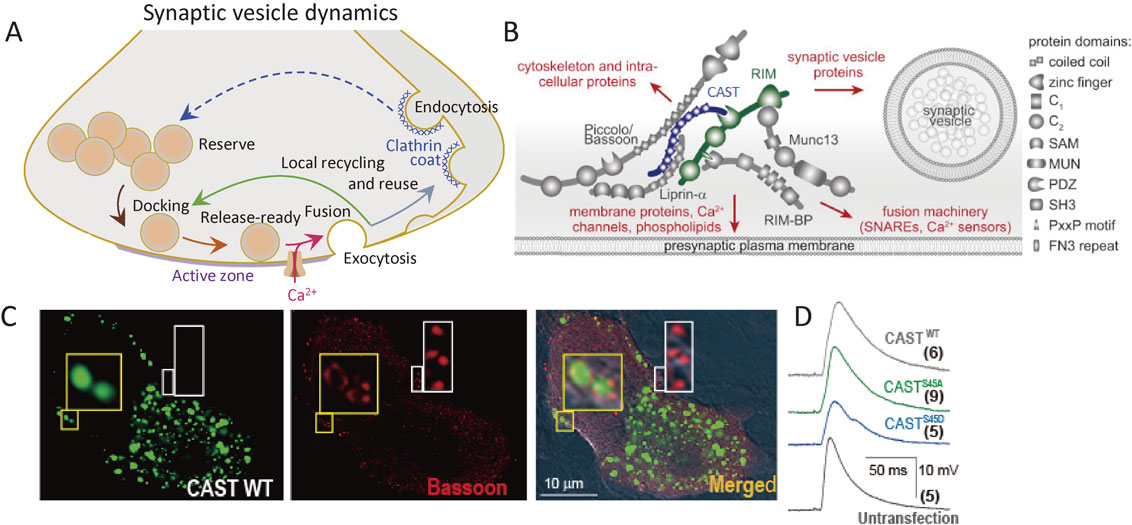
Synaptic vesicle (SV) dynamics, active zone (AZ) proteins in superior cervical ganglion (SCG) neurons, and change in the AZ with overexpression of cytomatrix at the active zone-associated structural protein (CAST). A, SV dynamics. SVs (orange circles) are loaded to the AZ, prepared to be release-ready, fuse with the presynaptic membrane upon Ca2+ influx, and then recycled. Adapted from Mochida, 2016.130) B, The AZ is a highly organized structure that docks SVs close to fusion machinery proteins (SNAREs), Ca2+ sensors, and Ca2+ channels. This establishes the tight spatial organization required for fast SV exocytosis upon Ca2+ entry, and it provides molecular machinery to set and regulate synaptic strength. Adapted from Wang S et al., 2016. C, CAST controls AZ architecture. Green fluorescent protein image with exogenous CAST (left), immunofluorescent image of Bassoon (center), and merged DIC image (right) of two SCG neurons; one expressing green dots 2 days after CASTWT transfection and another untransfected. Insets are enlarged images of boxed areas (×10). Yellow box shows co-localization of transfected CASTWT and Bassoon, while white box shows Bassoon colocalized with endogenous CAST (no image). The AZ architecture can be monitored by Bassoon immunofluorescent signals (red). Punctate Bassoon signals are seen in normal synaptic boutons (white box), whereas CAST overexpression resulted in a donut-shaped distribution of Bassoon (yellow box). D, EPSP waves recorded from untransfected postsynaptic neuron. The presynaptic neurons were transfected with CASTS45 DNA 2 days before the recording. As a control, an averaged EPSP wave, evoked by an AP in an untransfected presynaptic neuron, is shown (Untransfected). The number of averaged neuron pairs is indicated in parentheses. Adapted from Mochida et al., 2016.
The AZ is a highly organized structure that serves as a platform for SV exocytosis, mediated by SNARE proteins complex, and nearby Ca2+ channels (Fig. 5A, B). This arrangement establishes the tight spatial organization required for fast SV fusion upon Ca2+ entry and sets the synaptic strength.70) Although the full molecular composition of the AZ is unclear, many AZ proteins have been identified, including RIM, RIM-BP, Munc13, Bassoon, Piccolo, Liprin-α, and cytomatrix at the active zone-associated structural protein (CAST) (Fig. 5B).66),71)–77) These are all relatively large proteins with significant domain structures that interact with each other, forming a large macromolecular complex.78)
In 2002, my collaborator Toshihisa Ohtsuka discovered an AZ protein and named it CAST.74) Bruchpilot, a Drosophila ortholog of CAST, is required for AZ structural integrity and Ca2+ channel clustering,79),80) suggesting a potential function of CAST in the mammalian AZ.81) Indeed, CAST knockout in mice reduced retinal AZ size82) and impaired inhibitory neurotransmission in the hippocampus.83) We demonstrated that CAST is a core protein of the large macromolecular complex (Fig. 5B), and disruption of its interaction with Bassoon or RIM impairs synaptic transmission.66) Removing RIM,66) a key protein for synaptic vesicle dynamics,84),85) abolishes SV docking, reduces SV release probability, and reduces the speed of exocytosis.70) In addition, impairment of Bassoon slowed down SV reloading.86) Several lines of evidence indicate that the CAST complex plays fundamental roles in setting the synaptic strength. Therefore, we further examined CAST and CAST-related AZ protein functions, as described below.
3.2.2 Phosphorylation of AZ proteins (background to this study 2).The AZ serves as a site for preparing SVs to be ready for exocytosis. These SVs are termed the readily releasable pool (RRP).87) The size is controlled by SV recruitment to the RRP, and protein kinases activated by residual Ca2+ of AP bursts change the RRP size.88),89) Recent genomic and proteomic analyses have revealed more than 600 protein kinases in humans.90) Among the numerous presynaptic kinases, protein kinase C plays a role in SV recruitment88),91) and in a short-term form of presynaptic plasticity.92)–94) Protein kinase A is also involved in presynaptic long-term plasticity at cerebellar95) and hippocampal synapses.96),97) RIM has been proposed as the substrate of the phosphorylation events.98),99) However, later studies of a mouse line with a point mutation in the protein kinase A phosphorylation site of RIM, showing normal presynaptic protein kinase A-dependent long-term plasticity, suggested the presence of additional protein kinase A AZ protein substrate(s) that determine long-term presynaptic plasticity. These protein kinase substrate proteins may constitute a ‘phosphoswitch’ that determines synaptic strength for a while after AP bursts.
Bassoon also regulates SV recruitment to the release site during AP bursts,86) and may be target of AZ kinase(s). A candidate for the AZ kinase is an AZ-associated serine/threonine kinase, SAD-B. Collaborating with Toshihisa Ohtsuka, we demonstrated that SAD-B phosphorylates RIM.65) In addition, SAD-B interacts with CAST, a core of the complex of AZ proteins, including RIM (Fig. 5B).66) Loss of SAD-B from the complex by perturbing the interaction with CAST inhibited synaptic transmission and reduced the number of SVs in the RRP,65) suggesting that SAD-B may phosphorylate CAST. Therefore, we examined the phosphorylation of a candidate protein, CAST, in the rat brain synapses,18) and found that phosphorylated CAST is tightly associated with AZs. In vitro experiments showed that SAD-B phosphorylates CAST at its N-terminal Ser (S)-45 residue. Furthermore, increasing neural activity generating APs burst in SCG neurons facilitated CASTS45 phosphorylation.18)
3.2.3 CAST determines AZ architecture and the EPSP wave form.I microinjected SCG neurons with a phosphomimetic-CASTS45D DNA (phospho-Ser-45), serine (S) is substituted by the negatively charged aspartic acid (D), which partially mimics a phosphorylated protein) prepared in the Ohtsuka laboratory. Wild-type CAST was overexpressed as a control. Surprisingly, CAST overexpression increased the size of the AZ (Fig. 5C),18) by 4-fold compared with the normal AZ. In contrast, AZ expressing phosphomimetic-CASTS45D showed a lower density of other AZ proteins, such as Bassoon. These changes in AZ architecture suggested that CAST regulates AZ formation by the control of AZ protein distribution, and thereby determines the efficacy of transmitter release. Indeed, CAST overexpression prolonged the EPSP duration and increased the integral by 1.7-fold, because of an increase in the number of SVs in the RRP,100) whereas the EPSP peak amplitude was unchanged (Fig. 5D). In contrast, phosphomimetic-CASTS45D expression reduced the peak amplitude and the integral of the EPSP by about half (Fig. 5D), and reduced SV numbers in the RRP.18) These results suggested that CAST controls the RRP size but not SV release probability, and that the CASTS45 phosphorylation decreases the number of release-ready SVs.
3.2.4 CAST phosphorylation delays replenishment of the RRP.The speed of SV recruitment to the release site is monitored by the rate of recovery from the RRP depletion with AP bursts (Fig. 6A). Recovery of the RRP in SCG neurons consists of two phases: a fast component that corresponds to rapid reloading of the RRP with SVs from the reserve pool, and a slow component that is based on the gradual reloading of the RRP with SVs generated through endocytic pathways.19),101) Expressing CAST or CASTS45 mutants altered the recovery rate:18) CAST and phosphonegative-CASTS45A accelerated the fast recovery rate, whereas phosphomimetic-CASTS45D slowed it (Fig. 6A). Thus, CASTS45 phosphorylation decreases the RRP size by slowing down SV reloading during and after intense synaptic activity (Fig. 6C).
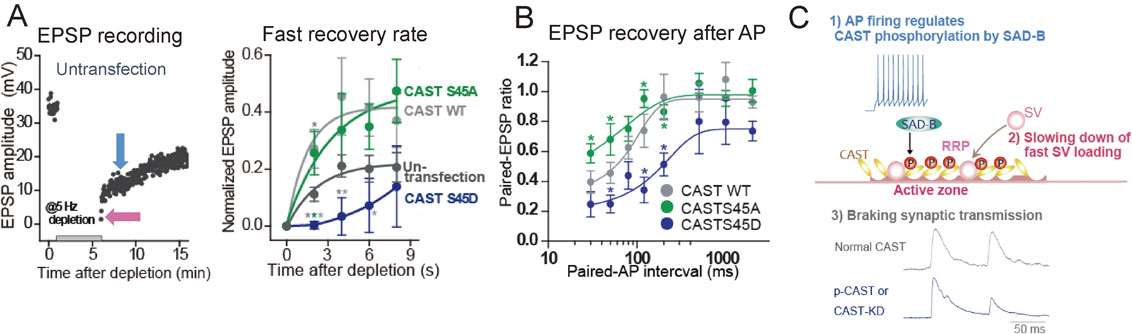
Recovery of synaptic vesicles (SVs) in the readily releasable pool (RRP) is accelerated by wild-type cytomatrix at the active zone-associated structural protein (CASTWT) overexpression and phosphonegative-CASTS45A, whereas it is delayed by phosphomimetic-CASTS45D. Presynaptic neurons were transfected with CAST or mutant CASTS45 cDNA 2–3 days before the excitatory postsynaptic potential (EPSP) recordings. A, EPSP recovery from depletion of SVs in the RRP. EPSPs were recorded every 2 s. A 5-min train of APs at 5 Hz was applied, as indicated, for depletion. EPSP amplitudes recovered with two different rates: fast (pink arrow) and slow phases (blue arrow) (left). The fast recovery rate was estimated by an exponential fit to the increase in EPSP amplitude from 0 to 8 s after the depletion (right). B, Phosphonegative-CASTS45A relieved the second EPSP reduction, whereas phosphomimetic-CASTS45D potentiated the reduction. The paired EPSP ratio was plotted against the paired-AP interval with SEM. *p < 0.05; unpaired Student’s t-test. C, Cascaded reactions of CAST phosphorylation. Adapted from Mochida et al., 2016.
Previous studies suggested that phosphorylation of AZ proteins induced by AP bursts lasts minutes.89) This is not the case with CAST phosphorylation. The time window for CAST phosphorylation estimated by the paired-AP protocol (Fig. 6B)18) revealed that a single AP induces CAST phosphorylation and the effect lasts 30–120 ms after the AP. Phosphonegative-CASTS45A decreased the paired-EPSP depression (<200 ms),19),101) suggesting that phosphorylated CASTS45 down-regulates SV reloading shortly after an AP, but not over longer (200 ms) time courses. In addition, phosphomimetic-CASTS45D enhanced the paired-EPSP depression. Thus, phosphorylation of CASTS45 negatively controls the RRP reloading within 200 ms of an AP firing (Fig. 6C). This ultrafast regulation of SVs reloading may be important for saving release-ready SVs for an incoming AP.
3.2.5 CAST knockdown mimics CAST phosphorylation.Although CAST knockout reduced the AZ size in the retina,82) no ultrastructural changes were observed in central synapses.83) The physiological significance of CAST phosphorylation could be examined by knockdown of CAST in presynaptic SCG neurons using shRNAs (short hairpin RNA produces continuously siRNA in the nucleus).18) Acute deletion of CAST by shRNAs showed no significant reduction in EPSP amplitude; however, the rate of the fast reloading phase after RRP depletion was significantly delayed by CAST deletion. Indeed, paired-EPSP depression was facilitated by CAST deletion. These results indicated that the inactivation of CAST by phosphorylation slows transmitter release when the next AP arrives at the presynaptic terminal shortly (<200 ms) after AP firing. This presynaptic regulation is reasonable for relatively low firing rate and slow signal conduction of sympathetic postsynaptic neurons.
3.3 Regulation of SV mobilizing to the release site — Activation of myosin IIB and VI —.Motor proteins activated by APs firing mediate translocation of SVs within presynaptic terminals.11) In 1994, I first demonstrated the role of neural myosin in the regulation of neurotransmitter release at presynaptic terminals11) where myosin IIB is highly expressed.102) After establishing a mammalian synapse for functional studies of presynaptic proteins,10) the first protein that I examined the role of in the presynaptic neuron was myosin II. Although its role in growth cone motility, but not SV mobilization, had been reported,103) I demonstrated that presynaptic AP firing activates myosin light chain kinase, and the resultant actin-myosin II interaction is involved in neurotransmitter release.11)
Twenty years later, live imaging of hippocampal neurons provided direct evidence that myosin II is required for SV motility during synaptic firing.104) The importance of myosin II in SV motility and synaptic transmission was also demonstrated at the Drosophila melanogaster neuromuscular junction,105),106) where myosin VI is also required for SV localization, short-term facilitation,107) and SV dynamics.108) In contrast, in brain synapses, myosin VI is postsynaptically involved in receptors transport.109)–111) Its role in SV motility at presynaptic terminals in the brain has not yet been elucidated, although myosin II regulates SV motility near the release site.112),113)
I have recently demonstrated that both myosin IIB and VI, activated by CaM sensing residual Ca2+, mediate ultrafast SV resupply to the transmitter release site. This section 3.3, after a brief introduction to actin and myosin in brain synapses, summarizes roles of myosin IIB and VI in distinct SV resupply pathways, studied with my students.12),101),114),115)
3.3.1 Myosin in brain synapses (background to this study).Myosins are a large family of actin-based cytoskeletal motors that use energy derived from ATP hydrolysis to generate movement and force.111) Myosin classes II,116) V,117) and VI118) have specific roles required for synapse function104),109),119) and several forms of synaptic plasticity.120),121) Their major roles at synapses are diverse and include the regulation of actin cytoskeleton dynamics in dendritic spines and powering of synaptic cargo transport.119)
The readily releasable SV cluster (i.e., the RRP) is thought to be filled up from a larger SV cluster, called the recycling pool or reserve pool, during sustained neural signals of APs (Fig. 5A).112) However, it is not yet understood how SVs are mobilized from one cluster to another. In 1994, I proposed that an actin-myosin II interaction is involved in SV resupply for SV exocytosis.11) Twenty years later, studies in the calyx of Held synapses and the cerebellar synapses of the parallel fiber and the molecular layer interneuron revealed that myosin II controls replenishment of the RRP.113),122) In these synapses, the RRP can be divided into two further pools, a fast-release and a slow-release pool.123) In response to AP bursts, myosin II converts slow-release SVs to fast-release ones122) with a rapid rate constant,113) suggesting that myosin II transport SVs in the RRP to the exocytosis site. However, how many nerve impulses are required for the activation of myosin II is still unknown. Thus, in the next section, I would like to discuss this question by studying the synaptic transmission of SCG neurons.
3.3.2 Myosin IIB and myosin VI activation by AP firing.Among three myosin II isoforms A, B, and C, myosin IIB and myosin VI are specifically expressed at presynaptic terminals of SCG neurons in culture.102),114) Myosin IIB is Ca2+/CaM-dependently activated by myosin light chain kinase, whereas myosin VI is directly activated by Ca2+/CaM. Participation of myosins IIB and VI in RRP resupply was monitored using the recovery kinetics of release-ready SVs with diverse AP firing patterns.11),114) Presynaptic terminals were microinjected with siRNAs to acutely reduce myosin IIB or VI [i.e., knockdown of IIB or VI (IIB-KD and VI-KD, respectively)]. Green fluorescent protein expressing cDNA confirmed the siRNA expression.
Can a single AP activate myosin? This question was answered by applying the paired-AP protocol (Fig. 7A).114) As described in section 3.2.4, SCG neurons show synaptic depression at short AP intervals (≤120 ms).12),20),115) VI-KD potentiated synaptic depression with <50 ms intervals (Fig. 7A). This means that a single AP activates myosin VI within 50 ms (Fig. 7D) and completes the resupply of release-ready SVs within 120 ms. In contrast, IIB-KD did not change the paired EPSP size, suggesting that a single AP cannot activate myosin IIB.

Myosin IIB and VI mediate synaptic vesicle (SV) resupply to the readily releasable pool (RRP). Presynaptic neurons were transfected with control, myosin IIA-, IIB-, or VI-siRNA 2–3 days before the excitatory postsynaptic potential (EPSP) recordings. A, Myosin VI, but not IIB, loss-of-function reduced release-ready SVs after action potential (AP)-evoked transmitter release. The averaged paired-EPSP ratio with SEM was plotted against the paired-AP interval. *p < 0.05; Bonferroni post hoc test after one-way ANOVA. Myosin IV knockdown (VI-KD) vs. control or myosin IIB knockdown IIB-KD (asterisks). B, Myosin IIB and VI loss-of-function impaired release-ready SVs during AP bursts at 10 Hz, resulted in gradual EPSP amplitude reduction. C, Myosin IIB and VI loss-of-function delayed recovery from RRP depletion. After a 1 min control recording at 1 Hz, a 4-min AP train at 5 Hz was applied to deplete SVs. EPSP amplitudes were normalized to the mean EPSP amplitudes before the 4-min train. Recovery rate of release-ready SVs was estimated by double exponential growth fit to the increase in averaged EPSP amplitude after depletion. +Dyn1: double knockdown of dynamin-1 and myosin; +Dyn3: double knockdown of dynamin-3 and myosin. D, Schematic drawing of SV resupply to the RRP by myosin VI and IIB through distinct SV recycling pathways mediated by dynamin isoforms. SVs clusters are classified into two pools: the RRP and reserve pool (RP). Adapted from Lu et al., 2009; Tanifuji et al., 2013; Mori et al., 2014; Hayashida et al., 2015.
The time window of myosin action is short, within hundreds of milliseconds. This was estimated by analyzing EPSP decreases after repetitive APs.114) Figure 7B shows that, with AP firing at 10 Hz, the second EPSP was more sensitive to VI-KD, suggesting that myosin VI resupplies SVs to the release site within 100 ms after an AP, which is consistent with the paired-AP protocol data (Fig. 7A). In contrast, the third EPSP was more sensitive to IIB-KD, suggesting that myosin IIB takes 200 ms or needs two APs for SV resupply. Actually, activation of myosin IIB, but not myosin VI, needs more frequent AP firing >0.1 Hz.114)
Where does myosin act? I would conclude that myosin IIB and VI contribute to local SV mobilization to the release site. Two pieces of evidence support this conclusion. With IIB-KD or VI-KD, (1) the number of release-ready SVs (≈50 SVs) was similar to that of controls (see first EPSP in Fig. 7B), (2) it was reduced severely after AP firing (Fig. 7B), but the size of the RRP calculated with the EPSP waveform was 7% or 14% less than those of controls.114)
3.3.3 Myosin IIB and VI reloading of the RRP through distinct SVs resupply pathways.Members of my research group have accumulated evidence that SVs are mobilized to the release site by myosin IIB and VI through distinct pathways (Fig. 7D).12),19),101),114),115) After AP bursts, RRP reloading with SVs showed two phases, fast and slow, due to different molecular contributions.101) The SV kinetics associated with myosin IIB and VI in the fast and slow RRP reloading phases were estimated using the recovery rate, applying the same depletion-recovery protocol as described in section 3.2.4 and Fig. 6A.114) The time constant for the fast recovery was τ = 8 s, and that for the slow recovery was τ = 4–5 min. IIB-KD moderated the fast recovery to τ = 14 s, but not the slow recovery (Fig. 7C). In contrast, with VI-KD, the fast recovery was faster, τ = 2 s, and the slow recovery was delayed. Furthermore, double knockdown with IIB-KD and VI-KD significantly delayed both fast and slow recoveries. Thus, the kinetic analysis suggested myosin IIB and VI specificity for the fast and slow SV resupply pathways (Fig. 7D).
After AP bursts, the fast and the slow SVs resupply pathways are mediated through distinct SV recycling pathways activated by dynamin-1 and -3,115) key proteins for plasma membrane retrieval known as endocytosis for SVs reuse.124) Double knockdown of dynamin-1 or -3 and myosin IIB or VI confirmed the contribution of individual isoforms to the fast and slow components (Fig. 7C). I would conclude that myosin IIB supports fast SV loading close to the release site through a dynamin-1-mediated SV recycling pathway, whereas myosin VI supports slow SV loading through a dynamin-3-mediated pathway. Both pathways reload a common SV pool dependent on Ca2+ signals accompanying neural activity of AP firing (Fig. 7D).115)
At fast synapses, presynaptic proteins implicated in SV exocytosis acts within milliseconds. However, the time order of other proteins implicated in the regulation of transmitter release has been unclear. With electrophysiological measurements of synaptic transmission from synapses where multiple presynaptic protein functions have been genetically manipulated, my studies investigated the time course of protein implication induced by residual Ca2+ ion dynamics, due to Ca2+-buffering near the SV release site. After firing of a single AP, several proteins are implicated in the completion of activity within a few hundred milliseconds (Figs. 4A, 6B, 7A and 8); this is a reasonable time for preparing SVs for the next round of transmitter release.

Millisecond Ca2+ dynamics activate multiple protein cascades for synaptic vesicle control. Myosin IIB and VI activated by Ca2+/calmodulin (CaM) dependent on action potential (AP) firing transport synaptic vesicles (SVs) for the readily releasable pool (RRP) recovery. In contrast, AP firing induced phosphorylation of cytomatrix at the active zone-associated structural protein (CAST) slows RRP replenishment. Elevation of Ca2+ with single AP firing activates Ca2+/CaM/myosin VI and the serine/threonine kinase SAD-B at the active zone (AZ), and controls the speed of SV reloading into the RRP (upper scheme), resulting in modulation of transmitter release efficacy for an incoming AP (bottom left traces). A train of AP firing controls the efficacy by myosin IIB activation (upper scheme) and possibly by other proteins in the AZ for the regulation of non-stop synaptic transmission (bottom right traces).
Live imaging of fluorescently labeled SVs has been developed for studying presynaptic protein functions in neurotransmitter release.125),126) The resolution time for fluorescent image is, however, insufficient for analyzing the time courses of presynaptic protein activity induced by a single AP. In contrast, electrophysiological measurements provide real-time monitoring of the signaling events occurring during synaptic transmission. Within hundreds of milliseconds, after spatial and temporal change in Ca2+ concentration, the Ca2+ ion dynamics activate Ca2+-binding proteins as a functional protein switch for transmitter release. Although biochemical studies suggest that phosphorylation reactions take minutes, my studies have found that phosphorylation of CAST by SAD-kinase controls the speed of SVs loading to the release site for 100 ms after a single AP, and myosin II activated by myosin light kinase mobilizes SVs 200 ms after a single AP (Figs. 6–8).
Classical physiological studies have demonstrated that presynaptic short-term plasticity in the facilitation of transmitter release is induced by the residual Ca2+ of a high frequency AP burst.127) As described in this review, residual Ca2+ ions after a single AP also control the efficacy of SV release because of the direct regulation of Ca2+ channels by CaM and neuronal CaS. Classical physiological studies also demonstrated that presynaptic depression is due to use-dependent depletion of SVs.2) However, my studies revealed that, in addition to SVs loss, the depression is mediated by Ca2+ channel regulation. Furthermore, for the non-stop ultrafast transmitter release events, neural activity selects a specific time window for depression and facilitation of transmitter release; this is mediated by the control of Ca2+ channel activity with CaM- and neuronal CaS-binding (Fig. 4), slowing SV replenishment by AZ protein phosphorylation (Fig. 6C), and activation of myosin IIB or VI114) in distinct dynamin isoform-mediated SV recycling pathways (Fig. 7D).113) However, most Ca2+-dependent reactions occurring in the presynaptic terminal are still remain to be elucidated.
Since a number of presynaptic proteins were identified in the early 1990s, the study of their function has been greatly advanced using live imaging techniques, visualizing SVs125),126) and protein interactions128),129) with fluorescently labeled SVs and proteins. These imaging techniques for SV visualization detect slow movement with non-physiological strong stimulation, but these disadvantages are gradually being addressed. Using fluorescence resonance energy transfer and fluorescence lifetime imaging microscopy,128),129) real-time imaging of presynaptic proteins is an attractive new approach to elucidate the molecular mechanisms of protein complexes for the regulation of neurotransmitter release.

Sumiko Mochida was born in Nagano Prefecture in 1952. She graduated from the School of Pharmacy, Kitasato University in 1975. She completed her Ph.D. as a research fellow at Tokyo Medical College in 1982, and then studied for 2 years as a postdoctoral fellow at the School of Medicine, University of California San Francisco, where she had been trained in electrophysiological measurement of synaptic transmission in cat, rabbit, and rat sympathetic ganglia. She returned to Tokyo Medical College as an assistant professor in 1984 and started to culture adult sympathetic neurons. Between 1988 and 1990, she joined the Centre National de la Recherche Scientifique where she studied the molecular mechanism of transmitter release blockade after genetic expression of tetanus and botulinum neurotoxins in Aplysia synapses. Immediately after her return to Tokyo Medical College, she established a useful mammalian synapse model with sympathetic neurons in culture. She has involved in collaborations with molecular biologists in the US, Europe, and Japan, and has elucidated the functions of numerous presynaptic proteins. Between 1996 and 1999, she received a Human Frontier Science Program Research Grant as a member of an international collaboration with five researchers in the US, France, and Germany. In 1999, she received the Saruhashi Prize, which is awarded to the leading woman scientist every year. During the research work at Tokyo Medical University she became an Associate Professor in 1997, a Professor in 2001, and a Visiting Professor of the Tokyo University of Pharmacy and Life Science (2005–), Kanazawa University (2006–2012), Shanghai Jiao Tong University (2008–2011), and National Institute of Physiological Science (2008–2012).
The author expresses her sincere thanks to all the collaborators who have participated in the studies described in this review.