Abstract
Glycosidic compounds are indispensable molecules in living systems. Biological phenomena such as cell wall formation, energy storage, and cell recognition strongly depend on the multi-functional characters of these substances. Development of highly regio- and stereoselective glycosylation reactions is necessary to provide sufficient amounts of specific compounds in basic research as well as for applications in industry. This review presents an overview of chemical and chemo-enzymatic glycosylations that have been developed during my forty-year academic career in the field of glyco-science. In the course of these studies, several new concepts such as “Direct Anomeric Activation”, “Glyco-Process Chemistry” and “Glyco-Chemistry Cycles” have been established.
Introduction
Climbing big trees is, at any time of the era, one of children’s favorite things. However, everyone has the scary experience of nearly falling when jumping from a small branch to another, because it is difficult to move firmly from one branch to the next. Glyco-science can be thought of as being big trees in which “organic synthesis” is the tree fluid containing the many nutrients that circulate through its vascular system of a tree and creates new disciplines as branches (Fig. 1).1) Interdisciplinarity is one of the most important steps for advancing glyco-science, and is like jumping from one branch to another, where one has to be adept with acrobatics to keep the momentum to advance into new forests. This is one direction of synthetic research in carbohydrate chemistry.
There is, however, another direction that is also important. We should view the entire glyco-science as a beautiful dendritic structure. We should look where we step on the tree and re-acknowledge that a tree never grows without a big trunk and the roots that firmly support many branches and efficiently absorb nutrients, respectively. A vital point to realize is that a big trunk and roots can form only when a sufficient amount of tree fluid circulates. “Organic Synthesis” is not only the nutrients for the frontier region but it is used for maintaining the support of the tree.
Since 1976, I have consistently devoted my effort to finding new glycosylation reactions as basic and general tools for maintaining glyco-science.2) Most of these findings were made as a result of serendipities (lucky accidents). The present paper overviews how these discoveries were made during my forty-year research period.
1. Development of chemical glycosylation
1-1. Scientific background.3)
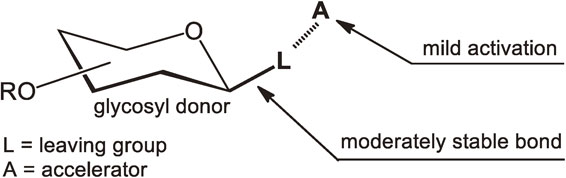
Since ancient times, human beings have used carbohydrates for daily necessities with textiles, starch, wood, paper, sweets, medicines, detergents being familiar examples. The main components of these compounds are glycosides that consist of a glycon part and an aglycon part connected through a glycosidic bond that can be created by a glycosylation reaction. Glycosylation is a process of reacting a glycosyl donor molecule and a glycosyl acceptor together. Selection of an appropriate glycosyl donor is of great importance in planning strategies for glycoside syntheses (Fig. 2). Factors to be considered concerning the structure of glycosyl donors are the chemical character of leaving groups (L in Fig. 2), the stereochemistry of leaving groups and the stability of protecting groups (R in Fig. 2). The following two characteristics are preferred for the chemical glycosylations to be employed in practical glycoside syntheses. First, the covalent bond between the leaving group (L) and the anomeric carbon atom should possess a moderate thermal stability, because the use of an unstable glycosyl donor often results in difficulty in its handling as well as low reaction yields. Second, the accelerator (A in Fig. 2) that promotes glycosylation by cleaving the C–L bond must have a weak acidity so that acid-sensitive functional groups are not damaged during the glycosylation reactions.
One of the oldest and most popular glycosylations is the Koenigs-Knorr reaction4) which consists of the treatment of unstable glycosyl chloride or bromide with alcohol in the presence of heavy metal salts such as silver oxide and mercuric cyanide. It is clear that the use of unstable synthetic intermediates and heavy metal accelerators are not suitable from the viewpoint of process chemistry. Therefore, there have been strong demands for designing more stable glycosyl donors that do not require activation by heavy metal salts.
1-3. Dinitropyridyl (DNPy) glycosides —the first glycosyl donor prepared using the nucleophilicity of sugar hemiacetals—.
It was not until 1979 that a glycosyl donor was designed based on the nucleophilic attack of 1-hydroxy sugar 1 to an electrophile 2 (Fig. 3).5) The resulting glycosyl donor 3 possessed an O–C=N moiety (in red) that can be activated by an acid catalyst. The reaction was discovered by chance. Our first goal was to replace the chlorine atom of 2-chloro-3,5-dinitropyridine 5 by a fluorine atom using potassium fluoride in the presence of a 1-hydroxy sugar 4 (Fig. 4). Instead of obtaining the required fluorinated dinitropyridine derivative, what we observed was an unexpectedly rapid substitution reaction by the 1-hydroxy sugar 4. The reaction completed within a few minutes at room temperature, affording 3,5-dinitropyridyl α-glucoside 6 in good yield. Later, these phenomena could be explained by a great enhancement of the nucleophilicity of the 1-hydroxy sugar 4 by the action of fluoride ion.
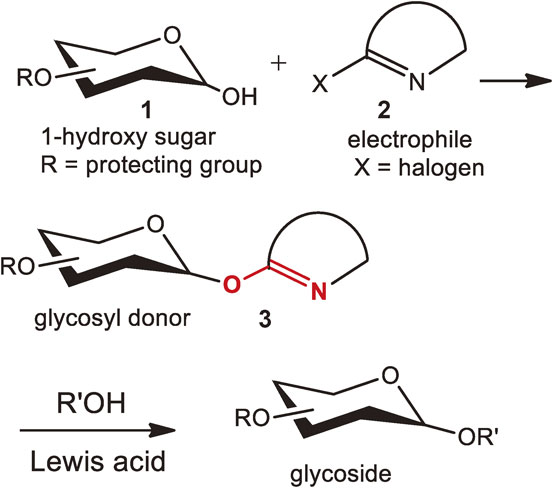
The resulting glycosyl donor 6 possesses an O–C=N moiety (in red) which can be activated by BF3-OEt2 in the presence of an alcohol, giving rise to the corresponding β-glucosides 7 in excellent yields. In 1980, Schmidt et al. improved the “imidate method” developed by Sinaÿ et al. in 19776) to the “trichloroacetimidate method” where an O–C=N moiety could be constructed using the nucleophilicity of hemiacetals and activated by a protonic acid or BF3-OEt2 (Fig. 4, lower-side).7)
1-4. Glycosyl fluoride method.8)
Fluorine has the smallest covalent radius among the halogens and the largest electronegativity among all elements. Because of the large bond-dissociation energy of the C–F bond (552 kJmol−1), it had been believed that glycosyl fluorides (L = F in Fig. 2) were too stable to be used for glycosidic bond formations. These beliefs influenced synthetic chemists for long time and resulted in glycosyl fluorides playing a less significant role than other glycosyl halides in carbohydrate chemistry, until the first publication of the glycosyl fluoride method appeared in 1981 (Fig. 5).9)

Once it was disclosed that the C–F bond of glycosyl fluorides could be activated by a weak Lewis acid, stannous chloride (A = SnCl2 in Fig. 2), the large bond dissociation energy of the C–F bond became inconsequential. On the other hand, the following merits of using glycosyl fluorides as glycosyl donors have emerged: a) ease of preparation under a variety of mild reaction conditions,10) b) relatively high stability toward chromatography on silica gel, c) variety of suitable promoters available for coupling with glycosyl acceptors,11)–14) d) orthogonality of the conditions required for activation of glycosyl fluorides over other glycosyl donors, for example, thioglycosides,15) and e) application to “armed-disarmed concept”16) for convergent synthesis of oligosaccharides.
1-5. How was the glycosyl fluoride method discovered? —Weak Lewis acid cleaves C–F bond—.
The discovery of the glycosyl fluoride method originates from two unexpected findings made during early experiments. The first finding was the realization of a new synthetic route to glycosyl fluorides by using a dehydrating reagent, 2-fluoro-1-methylpyridinium tosylate (8) (Fig. 6). In the course of investigating nucleoside synthesis using a dehydrating reagent, glycosyl fluoride was formed as the by-product in good yield starting from the hemiacetal. In the early 1980’s, the synthesis of glycosyl fluorides was a problem because preparative methods usually included the use of hazardous anhydrous hydrogen fluoride. Since the fluorination reaction using 8 required no hazardous reagents, this method triggered extensive investigations of glycosyl fluorides as glycosyl donors.

The second unexpected finding occurred during the course of examining various Lewis acids as potential accelerators for the coupling reaction of glycosyl fluorides and alcohols. Interestingly, in spite of the large bond-dissociation energy of the C–F bond, the desired glycosylation reaction proceeded smoothly with SnCl2, a fairly weak Lewis acid at room temperature, giving rise to 1,2-cis-glycosides that are difficult to prepare compared to the 1,2-trans-glycosides. When the reaction was carried out in the presence of silver perchlorate as a co-accelerator in diethyl ether, the 1,2-cis-selectivity further increased (Fig. 5). This method was found to be applicable to glycofuranosyl (five-membered) fluorides, affording a good route to 1,2-cis-furanosides that are difficult to obtain by other means.10)
The characteristic feature of the divalent tin compounds is that they have both a vacant orbital and a lone pair of electrons. In this glycosylation reaction, it is assumed that SnCl2 behaves as a Lewis acid where the vacant orbital accepts one of three lone pairs on the fluorine atom of the glycosyl fluoride. As a result of this interaction, the C–F bond cleaves to give the oxocarbenium ion intermediate that is then attacked by an alcohol to give the glycoside.
It is noteworthy that the addition of strong Lewis acids is not necessary to promote the glycosylation; the use of relatively weak Lewis acids like SnCl2 is sufficient to activate the considerably strong C–F bond of glycosyl fluorides. All of these results clearly show that the fluorophilic character of Lewis acids is much more important than their Lewis acidities. These basic findings have greatly stimulated the development of a series of new glycosylation reactions in these years, where a variety of combinations of glycosyl fluorides and Lewis acids have been designed on the basis of the fluorophilicity of the elements of the Lewis acids (Fig. 7). Among these promoters, tin(II) species,9) bis(cyclopentadienyl)metal derivatives (Cp2MCl2, M = Zr, Hf),14) BF3-OEt2,12) and protic acids17) are the most frequently employed.

The reason why glycosyl fluorides have been so frequently employed as glycosyl donors is that they show high activities in the presence of various promoters as well as ease of handling due to their thermal stability. Almost all elements that can activate the C–F bond of glycosyl fluoride donors are located in the region of hard elements with higher fluorophilicity in the periodic table (Fig. 7). It can be easily predicted that other Lewis acids that promote glycosylations using glycosyl fluorides will be found in the future based on the concept of fluorophilicity.
1-6. Application to natural product synthesis.
Since the reaction proceeds under mild reaction conditions, this original accelerator, SnCl2-AgClO4, was utilized by many researchers in the synthesis of complex glycosides such as Avermectin,18) Trimeric Lex,19) Agelagalastatin,20) β-Mannoside,21) and glycosphingolipid KRN7000 (Fig. 8).22) For example, the total synthesis of agelagalastatins, an antineoplastic glycosphingolipid, has been achieved by using an α-selective glycosylation of the ceramide derivatives with the trisaccharide fluoride promoted by SnCl2-AgClO4.

The glycosyl fluoride method, which was developed in the early 1980’s, has now reached a stage at which it has become possible to produce extremely complex glycosides while controlling their stereochemistry. Although the history of glycosyl fluoride method is much shorter than that of the classical Koenigs-Knorr glycosylation, many applications to natural product synthesis have already been demonstrated, clearly indicating its utility in the field of carbohydrate chemistry.
1-7. Protection-free glycosylation of oligosaccharides —In the margin of an Emil Fischer work—.
Supplying simple alkyl glycosides is an important role of a synthetic organic chemist, because these compounds can create their own demands as useful building blocks for construction of complex glyco-materials. The most simple and direct route for synthesis of unprotected alkyl glycosides was first reported by Emil Fischer in 1893.23) Emil Fischer (1852–1919) was the greatest of all organic chemists and made extraordinary contributions to glyco-science. After the monumental work on the establishment of the stereochemical configuration of all aldosugars, Fischer extended his studies to glycoside synthesis.
An unprotected monosaccharide was condensed with an alcohol in the presence of HCl catalyst usually at elevated temperatures, resulting in the net loss of water and substitution at the anomeric position by the alcohol. Fischer glycosylation discovered at the end of 19th century has, over the last century, remained one of the most popular methods due to its simplicity and versatility. The reaction proceeds without protection of the hydroxy groups. Despite the great utility for carbohydrate synthesis, the use of a strong acid has hampered the applicability of Fischer protocol to oligosaccharides, which Fischer pointed out himself in the original paper as follows.
“Dagegen scheinen die beiden Hexobiosen, welche noch eine Aldehydgruppe enthalten, der Milchzucker und die Maltose, fuer das Verfahren nicht geeignet zu sein, weil sie unter den Bedingungen des Versuches durch die starke Salzsaure gespalten werden.”
(It appears that the procedure is not suitable for disaccharides such as lactose and maltose because they are cleaved by hydrogen chloride.)
For the case of employing oligosaccharides as starting sugars, acetal exchanges of the inner glycosidic bonds would take place as a result of intermolecular protonations by the acid catalyst to the glycosidic oxygens (Fig. 9(A)). In fact, when melibiose, which is a disaccharide having an acid-labile 1,6-glycosidic bond, was treated with methanol in the presence of hydrogen chloride under Fischer’s conditions, the product was a mixture of monosaccharide derivatives as a result of glycosidic bond cleavage.
To avoid these acid-mediated side interactions, a site-specific intramolecular activation at the reducing end (direct introduction of the X∪Y–Hδ+ moiety in Fig. 9(B)) is indispensable. However, such a chemical technique has not been realized so far because the regioselective modification of the reducing end of unprotected sugars is extremely difficult. The development of a new methodology for specific anomeric activation of unprotected sugars has, therefore, been under demand.
In 2013, we developed a convenient protection-free synthetic route toward alkyl glycosides.24) In the course of our studies for developing a protection free glycosylation by using one-step preparable glycosyl donors, we were interested in the chemical behavior of 4,6-dihydroxy-1,3,5-triazin-2-yl β-glucoside and tried to prepare it by hydrogenolysis of the corresponding 4,6-dibenzyloxy derivative as a synthetic precursor. Unexpectedly, instead of obtaining the desired compound, we observed a quantitative formation of methyl α-glucoside (Fig. 10). These results suggested that our expected product was too reactive to be isolated and was immediately attacked by methanol that was used as solvent for hydrogenolysis, giving rise to the corresponding α-glycoside with inversion of configuration at the anomeric carbon atom.
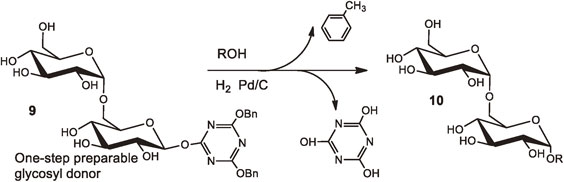
The alcoholysis of glycosyl donors, 4,6-dibenzyloxy-1,3,5-triazin-2-yl (DBT) glycosides 9, under hydrogenolytic conditions gave the corresponding α-glycosides 10 in good yields without adding any acid promoters. The method could be successfully applied to the conversion of acid-labile melibiosyl derivative 9 to the corresponding methyl α-melibioside 10. It is noteworthy that the present method can be applied to oligosaccharides, which has long been difficult to realize by the classical Fischer protocols. Alkenyl glycoside and alkynyl glycosides could also be prepared by using triethylsilane instead of hydrogen as a reductant. The present method could pave the way towards the glycosylation of acid labile oligosaccharides as well as acid labile aglycon alcohols.
2. Development of chemo-enzymatic glycosylations based on substrate design of glycosyl donors
2-1. Historical background.3)
All of the catalysts or accelerators employed for chemical glycosylation reactions are in great debt to the progress of organic chemistry, inorganic chemistry and organometallic chemistry in its long history where many types of catalysts have been prepared by chemical reactions in a test tube. Let’s turn our topic into fermentation, another activity of human beings for production of enzymes as macromolecular catalysts. The broad definition of fermentation is to transform organic substances into other useful compounds by the action of enzymes that are produced by microorganisms such as molds, yeasts, and bacteria.
Since ancient times, human beings have used fermentation for making bread, cheese, wine, and other food products without understanding the existence of the microorganisms involved. After several breakthroughs in the second half of the 19th century including Louis Pasteur’s discovery25) that living microorganisms cause fermentation, in 1897 Eduard Buechner finally succeeded in extracting a juice from yeast that ferments a sugar solution, affording alcohol and carbon dioxide.26) Triggered by these discoveries, fermentation technology advanced by mutating the microorganisms with physical and chemical treatments.
Based on the great progress of the two fields having a different cultural background, namely chemistry and biology, researchers of the 21st century took advantage of their accumulated knowledge and technologies of catalysts and prepared a wide variety of useful organic compounds.
2-2. Why are enzyme catalysts necessary for construction of oligo- or poly-saccharide structures?
Of the large variety of ways in which monosaccharide building blocks can be linked together, apparently only one leads to success. Glycosidic bond formation is achieved by a chemical reaction between the anomeric carbon atom (C1) of one monosaccharide molecule and a hydroxy group of another monosaccharide molecule, and results in an oxygen bridge between the two molecules. However, there are several alternatives for this reaction. A monosaccharide has five hydroxy groups attached to carbon atoms C-1, C2, C3, C4 and C6, and any one of them can undergo reaction with the C1 carbon of the other monosaccharide molecule, yielding what we may call a 1–1, 1–2, 1–3, 1–4, or 1–6 bond. In construction of the oligo-saccharide backbone, only one glycosidic bond must be formed. Such a selective bond formation is called a regioselective reaction.
In addition to achieving regioselectivity, synthetic chemists must deal with another ambiguity in the formation of a glycosidic bond. The hydroxy group at the anomeric carbon atom of, for example, d-glucose can point either downward or upward, giving rise to the α and β-isomers of d-glucose, respectively. Hence, two types of glucosidic bond can also be formed, the α-glucosidic bond pointing downward and the β-glucosidic bond upward.
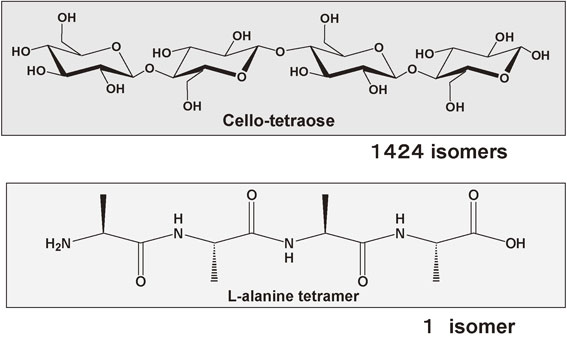
By considering all stereochemical alternatives, eleven isomers can be produced when a disaccharide is formed, 120 for a trisaccharide, and 1424 for a tetrasaccharide (Fig. 11, upper part).27) It is interesting to compare this with the fact that only one isomer is possible for l-alanine oligomers regardless of the degree of polymerization (Fig. 11, lower part). For an oligo- and poly-saccharide chain with several hundreds or thousands of mono-saccharide units, the number of isomers becomes astronomical. The difficulty in cello-tetraose synthesis originates from the requirement to achieve such a complete selectivity in all glycosylating reactions involved. These considerations indicate that we must achieve one particular configuration among 1424 possibilities. Synthesis of an oligosaccharide in a test tube had, therefore, been a challenge for synthetic chemists for long time.
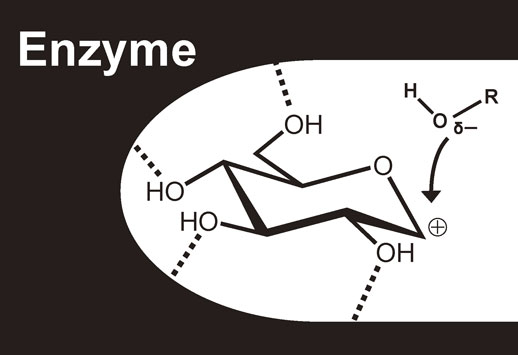
Glycosidases show perfect selectivity in transglycosylation reactions, giving rise to only one isomer because the formation of undesired isomer becomes impossible due to the steric hindrance caused by the wall of amino acid residues in the active site of enzyme (Fig. 12). For example, in case of β-glucosidase, the attack of the hydroxy group takes place from the β-face of the glucose, leading to stereoselective formation of a β-glucosidic bond.
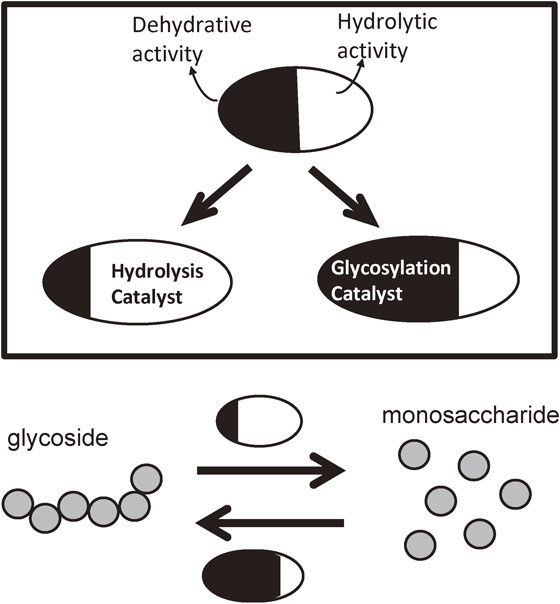
Glycosidase-catalyzed dehydrative condensations can be regarded as the reverse reaction of the hydrolytic process catalyzed by glycosidases. According to the principle of microscopic reversibility, glycosidases can catalyze not only hydrolysis but also the reverse reaction, dehydrative condensation, through the same transition state. The use of glycosidases as catalysts for producing glycosidic compounds is, therefore, commonly used in modern synthetic chemistry and engineering and has lead to the creation of new chemical industries that contribute to our society from the viewpoint of green and sustainable processes. Glycosidases possess two complementary activities, dehydrative activity and hydrolytic activity (Fig. 13). In nature, the role of glycosidases is to hydrolyze the glycosidic bonds of polysaccharides. Under special conditions, glycosidases can catalyze a glycoside bond formation, giving rise to the corresponding glycosidic compounds. However, to achieve an efficient enzymatic glycosylation, it is absolutely important to introduce an appropriate leaving group on the anomeric carbon atom.
Since it was found that a glycosyl fluoride derivative is recognized by a glycosidase, several studies on the enzymatic polycondensation reaction of glycosyl fluorides have been demonstrated. For example, the enzymatic polycondensation of β-cellobiosyl fluoride catalyzed by a cellulase has been reported.28) However, in these reactions, it was impossible to control the degree of polymerization. The glycosyl fluoride derivative behaved as glycosyl donor and as glycosyl acceptor at the same time, leading to the formation of a mixture of self-condensed products; a selective cross-coupling between the glucose moiety with another saccharide moiety could not be achieved. We postulated that, if the 4-OH group of the glucose could be protected by an appropriate water-soluble protecting group, an enzymatic cross-coupling would be possible provided the protected substrate was recognized by the cellulase effectively.
A convenient method for the preparation of cello-oligosaccharide has been developed which utilizes β-lactosyl fluoride as the glycosyl donor (Fig. 14).29) The reaction consists of the following enzymatic process: (1) a cellulase-catalyzed regio- and stereoselective lactosylation of cellobioside as the glycosyl acceptor, utilizing the transglycosylating ability of an enzyme-substrate complex formed from β-lactosyl fluoride and cellulase; (2) β-galactosidase-catalyzed cleavage of the terminal galactose unit from the lactosylated product, giving rise to a cellotriose derivative. By repeating these enzymatic reactions, cello-tetraoside derivative has been prepared in stereo- and regioselective manner. It should be noted that the 4-OH group of the glucose unit is protected by introducing a β-galactose moiety, avoiding self-condensation of the substrate.
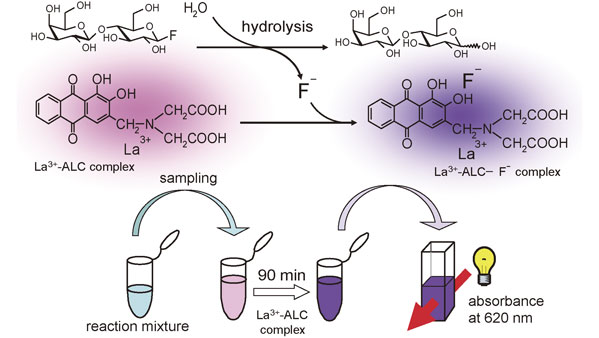
2-4. Fluoride ion-detecting enzyme screening (FIDES).
Finding an appropriate combination of glycosyl fluoride substrate and enzyme catalyst is key to designing an enzymatic glycosylation. The development of a facile procedure has been strongly required for the screening of enzymes that recognize glycosyl fluorides. We postulated that if the fluoride ion liberated from a glycosyl fluoride can be detected effectively in the presence of sugar compounds, enzyme proteins, and buffer components, a new enzyme assay would be available for evaluating the glycosyl fluoride-hydrolyzing activity of glycosidases. A novel colorimetric assay for glycosidases has been developed by using glycosyl fluorides based on the lanthanum–alizarin complexon (La3+–ALC) method (Fig. 15).30) In analytical chemistry, the La3+–ALC method is known to be one of the most reliable techniques to detect fluoride ion in aqueous solutions. The present method consists of the glycosidase-catalyzed cleavage of the C–F bond in a glycosyl fluoride and the subsequent complex formation of the liberated fluoride ion with La3+–ALC reagent. According to this method, endo-glucanase III which cannot be detected by the conventional method of using p-nitrophenyl glycoside could be detected with higher sensitivity by using glycosyl fluoride substrates (Fluoride ion-detecting enzyme screening (FIDES)).
“Serendipity”, a fortunate happenstance, sometimes occurs in our everyday research life. The term originates from Persian fairy tale, The Three Princes of Serendip (an ancient name for Sri Lanka), where three princes were always making discoveries by accident.
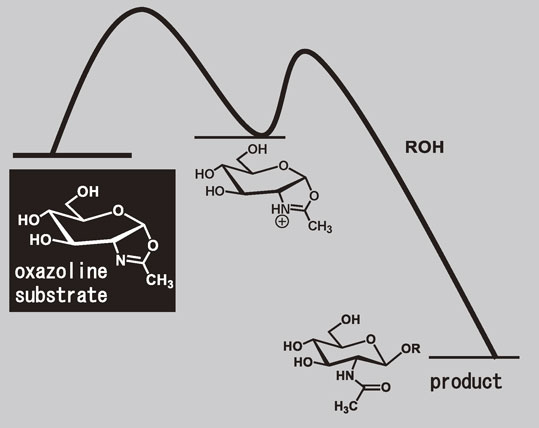
The enzymatic glycosylation by using sugar oxazoline-type glycosyl donors was disclosed by three coincidental fortunate happenstances. In Japan it has often been said that “A loach is not caught twice under the same willow tree” (You cannot always succeed twice running). Encouraged by the glycosyl fluoride chemistry described in the previous section, we just applied the procedure to N-acetylglucosamine on the shallow hypothesis that N-acetylglucosaminyl fluoride would also be an excellent glycosyl donor substrate for N-acetylglucosaminidase. However, the synthesis of desired N-acetylgluosaminyl fluoride failed. What we observed was an almost quantitative formation of sugar oxazoline derivative, the intramolecularly cyclized product of β-N-acetylgluosaminyl fluoride, as the result of neighboring participation of the acetamido group.
Although the student who was responsible for the experiment was very disappointed by the result, he became interested in the chemical behavior of the resulting sugar oxazoline. He saved it for the next enzymatic reaction, because he thought that it is a waste to throw it away with the Japanese “Mottainai” Spirit. Then, to prepare the first enzymatic reaction, he checked the pH of the aqueous solution of the sugar oxazoline with great disappointment again. The solution was too basic (pH ≈ 10) to be used for enzymatic reaction, presumably due to a small amount of NaOH that contaminated the sample in the last step of the synthesis. Ever the optimist, he ran the enzymatic reaction anyway, using the reaction mixture containing what he knew to be too drastic for the enzyme to be active. He was delighted and astonished to obtain an almost quantitative yield of polymerized product of artificial chitin, a polysaccharide containing N-acetylglucosaminide units.31) The success of the artificial chitin synthesis can be explained by the following three lucky coincidences.
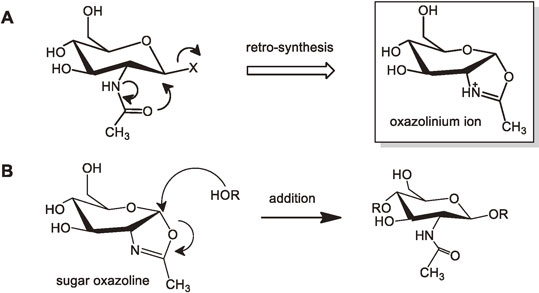
(a) Retro synthesis of N-acetylglucosaminide unit.
An N-acetylglucosaminide can be retro-synthetically disconnected to give an oxazolinium ion as a synthon (Fig. 16. A). On the basis of this retro-synthetic analysis, the real reaction in flask for construction of the N-acetylglucosaminide would be the addition of an alcohol to the anomeric center of a sugar oxazoline derivative (Fig. 16. B). In fact, sugar oxazolines are known to be a useful intermediate in the direct conversion of β-1-acetates to glycosides.32) Retro-synthesis is achieved by transforming a target molecule into precursor structures without assumptions regarding starting materials.33) What we obtained contrary to our expectation was a sugar oxazoline whose structure was almost the same as the oxazolinium ion; we could obtain the retro-synthetically rationalized starting material exactly by chance. This was the first happy accident for us.
The advantage of using sugar oxazolines is that the transglycosylation proceeds smoothly due to its lower activation energy. Sugar oxazolines are bicyclic molecules having a distorted structure where a six-membered pyranose ring and a five-membered oxazoline ring are fused sharing the anomeric and C2 positions. Consequently, sugar oxazolines possess higher potential energy compared with the conventional glycosyl donors such as p-nitrophenyl glycosides. The use of sugar oxazolines would, therefore, be a very promising for an efficient chemo-enzymatic glycosylation provided that such an artificial substrate with an oxazoline ring is recognized by a naturally occurring enzyme.
(b) Hydrolysis catalyzed by some kind of chitinases proceeds through an oxazolinium ion intermediate. —Substrate assisted catalyst—.
Chitinases hydrolyze the N-acetylglucosaminide unit of chitin, a β1→4 linked polysaccharide composed of N-acetylglucosamines. A novel hydrolysis mechanism of chitinases that involves an oxazolinium ion intermediate named “substrate-assisted catalysis” has been proposed by another group (Fig. 17).34) According to this mechanism, the glycosidic bond is cleaved as follows. First, the oxygen of the glycosidic bond is protonated by the carboxylic acid of an acidic amino acid followed by the formation of the oxazolinium ion intermediate by the nucleophilic attack of the amide carbonyl group to the anomeric center. The resulting oxazolinium ion intermediate is then attacked by water to give the hydrolyzate.

The sugar oxazoline we obtained could exactly be regarded as the transition state analogue for the above mentioned hydrolysis reaction catalyzed by chitinases. Some sort of chitinases may have acquired an extremely reasonable pathway for hydrolysis of an N-acetylglucosaminide moiety in the course of evolution. We were so lucky to have encountered an appropriate chitinase from the first experiment. This is the second happy accident for us.
(c) Too harsh conditions brought good results.
Enzyme activities are affected by changes in pH. The most favorable pH value where the enzyme is most active is known as the optimum pH (Fig. 18). Extremely high or low pH values generally result in complete loss of activity for most enzymes. The reason why the enzymes lose their hydrolytic activity is that catalytic residue, usually carboxyl acid acting as a general acid, is deprotonated under basic condition. Thus, this catalytic residue is not able to work as a general acid catalyst.
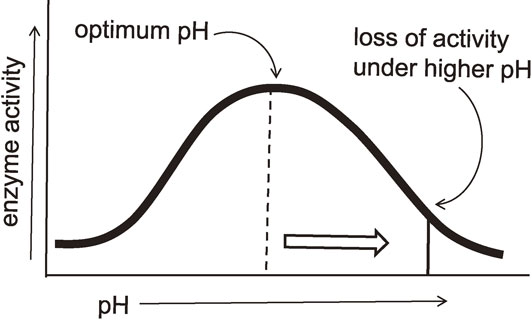
The optimum pH value for the chitinase is known to be in the range of 7–8. Our first experiment was performed under basic conditions (pH ≈ 10). Therefore, under the conditions of our first trial, the hydrolytic activity of the enzyme must have been greatly decreased. Nevertheless, the transglycosylation took place efficiently. The use of a transition-state analogue substrate allowed the reaction to proceed smoothly due to the small activation energy. In addition, the reaction proceeded only in the direction of transglycosylation because hydrolytic activity of the enzyme had been greatly decreased (Fig. 19). If the student had succeeded in adjusting the solution to the optimum pH, he would have never obtained the product, because the product would have already been hydrolyzed by an active chitinase. This is the third lucky accident.
2-6. A mutant N-acetylglucosaminidase enables efficient transglycosylations.
In the previous section, the chitinase-catalyzed trasglycosylations were performed under basic conditions to ensure the irreversibility of the glycosylating process. To improve the glycosylation reaction so that it can proceed under a different pH, especially under neutral conditions, it is necessary to decrease the activity of the enzyme by other methods. How to deactivate the enzyme? We selected the point mutation.
(a) Effect of mutations on hydrolysis activity of enzymes.
The enzymes employed were mutant chitinases, where the tryptophan residue 433 was changed to other amino acids. We tested whether the oxazoline derivative could be recognized by the active site of these mutant chitinases. When the substrate was kept at 37 °C in solution in a phosphate buffer (pH 7.0) without enzyme, only very slow hydrolysis (ring opening of the oxazoline moiety) was observed, indicating that the oxazoline is considerably stable under neutral conditions. Addition of the wild-type chitinase caused an extremely rapid hydrolytic ring opening of the oxazoline moiety. The mutant W433A (Fig. 20) gradually hydrolyzed, although W433A show no hydrolytic activity toward pNP-(GlcNAc)2. These results indicated that the distorted sugar oxazoline was able to be recognized by mutants while the mutant showed no hydrolytic activity toward an N-acetylglucosaminide moiety having no ring strain.
(b) Effect of mutations on the transglycosylating activity.
The effect of the mutation on the transglycosylating activity was investigated. Transglycosylation reaction was performed between a disaccharide oxazoline (Gal-GlcNAc-oxa) as glycosyl donor and GlcNAc-GlcNAc as a glycosyl acceptor at pH 7.0. The wild-type chitinase gave the corresponding tetrasaccharide (Gal-GlcNAc-GlcNAc-GlcNAc) in 70% yield after 2 min from the initiation of the reaction. However, the resulting product was rapidly hydrolyzed to trisaccharide and GlcNAc at prolonged reaction times and the yield became almost zero after 24 h. When the mutant chitinase (W433A) was used as catalysts, the yields after 24 h was 93%; the hydrolysis was suppressed due to the use of mutant chitinase with lower hydrolytic activities. This is the first example of enzymatic transglycosylation by the combined use of an activated substrate and a deactivated glycosidase.35),36)
2-7. Synthesis of oligosaccharide-containing thermosensitive polymers.
Saccharides have attracted much attention because of their potential as functions in biological recognition processes, and the applications of similar functional polymers are expected in the biomedical field. Glycopolymers are most commonly synthesized by either homo-polymerization of a sugar-containing vinyl monomer or by its co-polymerization with other polymerizable vinyl monomers. In conventional organic synthesis, multi-step protection and deprotection procedures are inevitable for introducing an oligosaccharide chain into the monomer.
Poly N-isopropylacrylamide (pNIPAM) which is one of the most studied thermosensitive polymers exhibited a rapid and reversible hydration-dehydration change in response to small temperature cycles around its lower critical solution temperature (LCST).37) Our interest was to know how thermosensitive polymers containing tri- and higher saccharides behave in aqueous solutions. No studies seem to have been reported on the topic probably because of the difficulty in introducing oligosaccharides into the polymer side chains.
We prepared chitooligosaccharide-containing monomers by using the following two enzymatic reactions. The first step involves a chitinase-catalyzed, highly selective transglycosylation of the Gal-GlcNAc oxazoline derivative to an oligosaccharide, giving rise to a new oligosaccharide having a galactose unit at the non-reducing end. The second step is a regioselective deprotection of the terminal galactose unit by the action of β-galactosidase, affording the corresponding oligosaccharides. As a result of these enzymatic processes, a GlcNAc unit can be added in a defined way to the non-reducing end of the starting oligosaccharide without accompanying self-coupling reactions. It should be noted that the galactose unit plays an important role as a suitable protecting group for the GlcNAc moiety in preventing the self-coupling reaction.
The resulting macromonomers can be copolymerized with NIPAM to give sugar-containing pNIPAM. Copolymerizations were performed in a distilled water to take advantage of the high solubility of the sugar-containing monomers in water. All copolymers were soluble in water at room temperature. The LCST of the aqueous solution of a pNIPAM with the Gal-(GlcNAc)2 side chain was determined to be 46.2 °C (Fig. 21). The sugar moiety in the side chain plays an important role in the liquid-solid phase transition as it can be seen that the LCST of the aqueous sugar containing pNIPAM solution dramatically increased.38) The advantage of the present procedure is that it is easy to obtain oligosaccharide-containing vinyl monomers without using any protection or deprotection procedures.
3. Direct anomeric activation in water —from glyco-chemistry to glyco-process-chemistry—
During the summer 1992–1995, I had several opportunities to visit Austin, Texas for a collaborative project with a plant physiologist on cellulose synthesis using glycosyl fluoride substrate. There, I found that it was very difficult for biologists to prepare the substrate by themselves. Consequently, I had to transport the substrate by myself with great attention and care. The synthesis of glycosyl fluoride substrate requires at least four steps from unprotected sugars, and is probably not so easy even for skilled organic chemists. These facts eventually inspired me to develop a more simple and practical method for glycosyl donor synthesis that could be used by many scientists who have little experience in organic synthesis.
In principle, glycosidase-catalysed transglycosylations require activated glycosyl donors such as p-nitrophenyl glycosides, glycosyl fluorides, or sugar oxazolines, the use of which greatly contribute to an increase in transglycosylation yields. The preparation of these glycosyl donors necessitates laborious tasks including the protection of all of the hydroxy groups, the regioselective introduction of a bromine or chlorine into the anomeric center under acidic conditions, the nucleophilic substitution of these halogen atoms by, for example, p-nitrophenol or fluorine atom, and the removal of the protecting groups (Fig. 22, below).
Therefore, development of a new class of glycosyl donors that can be prepared easily from the corresponding unprotected sugars was deemed necessary for establishing a practical chemo-enzymatic glycosylating process (Fig. 22, above). Our first question focused on whether it could be possible to activate the hemiacetal chemo-selectively.
3-1. DMT-glycosides are hydrolyzed by the corresponding glycosidases.
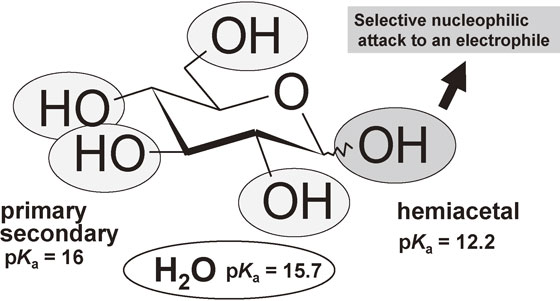
There are three kinds of hydroxy groups in a saccharide moiety, the hemiacetal, the primary hydroxy group, and the secondary hydroxy groups (Fig. 23). The pKa value of the hemiacetal is 12.2 whereas those of the primary and secondary hydroxy groups are around 16. In addition, the pKa of water is 15.7 in-between those values. This suggests that a chemo-selective nucleophilic attack of the hemiacetal becomes possible in water without protecting primary and secondary hydroxy groups, provided that an appropriate electrophile is chosen.
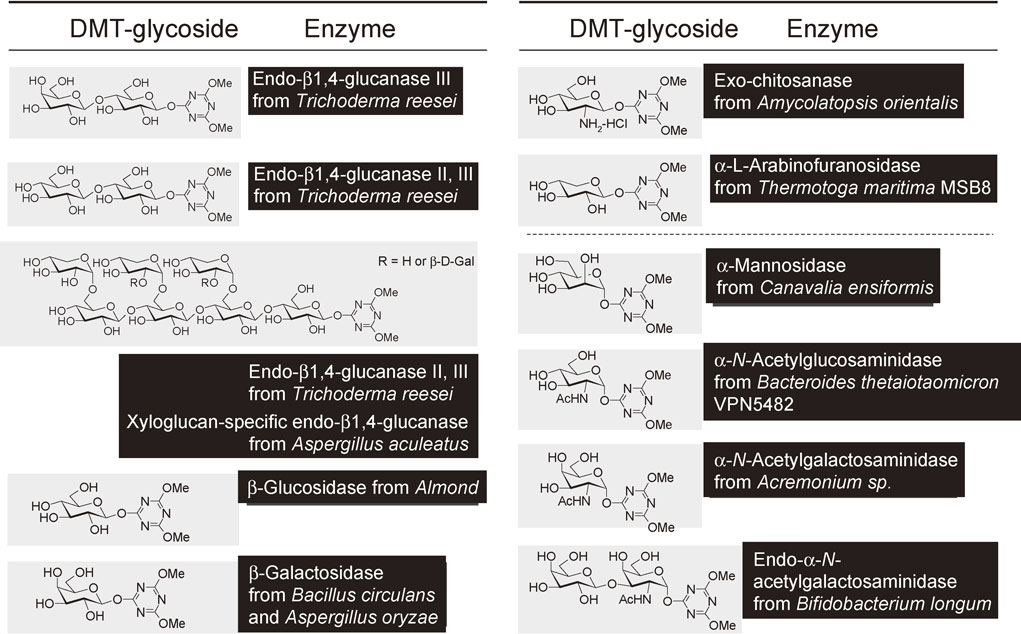
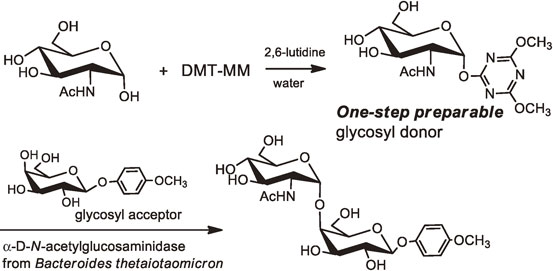
As a novel glycosyl donor for chemo-enzymatic glycosylation, we have developed 4,6-dimethoxy-1,3,5-triazin-2-yl glycosides (DMT-glycosides), which can be directly prepared from the corresponding unprotected sugars in water (Fig. 24 and Fig. 26). The resulting triazine derivatives were found to be recognized by various glycosidases and are able to be used as glycosyl donors for glycosidase-catalyzed transglycosylation reactions.
The new glycosyl donor substrates, DMT-glycosides, possess the following merits compared with conventional glycosyl donors such as p-nitrophenyl glycosides:
-
a.
synthesis of DMT-glycosides does not require protection of any hydroxy groups.
-
b.
synthesis can be done in aqueous media.
-
c.
DMT-glycosides are stable and can be stored at room temperature.
-
d.
DMT-glycosides can be well recognized by the corresponding glycosidases.
-
e.
transglycosylating activity of DMT-glycosides is higher than that of the conventional p-nitrophenyl glycosides.
The 4,6-dimethoxy-1,3,5-triazin-2-yl β-lactosides (DMT-β-Gal-Glc) have been prepared directly in water from lactose. The reaction has been carried out on a laboratory scale without protecting the hydroxy groups. The resulting triazine derivative was found to be recognized by endo-β1,4-glucanase III from Trichoderma reesei (EGIII). The EGIII-catalyzed transglycosylation of 4,6-dimethoxy-1,3,5-triazine derivative (DMT-β-Lac) with various glycosyl acceptors has successfully been demonstrated, affording the corresponding lactosylated products.39),40)
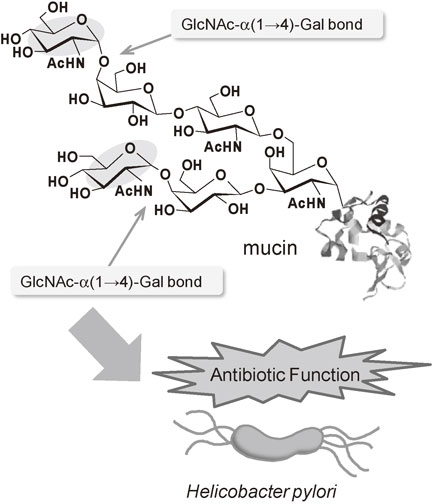
Oligosaccharides having an α-linked N-acetylglucosamine (GlcNAc) unit play biologically important roles in nature. The α-N-acetylglucosaminyl galactose (GlcNAcα1-4Gal), the terminal disaccharide unit of gastric mucin, is closely related to the infection of Helicobacter pylori in gastric mucosa (Fig. 25). Since compounds having the GlcNAcα1-4Gal unit would be expected as potential inhibitors in the growth of Helicobacter pylori, their efficient and practical synthesis in a stereospecific manner is necessary. We have established an efficient chemo-enzymatic process for construction of the α-linked disaccharide unit (GlcNAcα1-4Gal) found in gastric mucin (Fig. 26).41) The process consists of a one-step preparation method of a novel triazine type glycosyl donor in water and the subsequent transglycosylation to a galactose derivative catalyzed by α-N-acetylglucosaminidase.
3-2. Direct synthesis of sugar oxazolines in water.
The synthesis of sugar oxazoline derivatives necessitates a tedious process such as conventional protection/deprotection of hydroxyl groups. The chemical approaches involve careful design of protecting groups, catalysts and solvents. We discovered a direct method for synthesis of sugar oxazoline derivatives from the corresponding N-acetyl-2-amino sugars in aqueous media by using a chloroformamidinium type dehydrating reagent.42),43)
The intramolecular dehydrative reaction between the 2-acetamido group and the anomeric hydroxy group of unprotected N-acetyl-2-amino sugars takes place smoothly in water, leading to the formation of a 1,2-oxazoline moiety in good yield. The present method could be successfully applied to chito-oligosaccharides, saccharides with acid functions, and a complex-type oligosaccharide derived from a glycopeptide.
Chemo-enzymatic glycosylating process consists of the following two steps: 1) preparation of a glycosyl donor, and 2) transglycosylation to an glycosyl acceptor catalyzed by an enzyme. Since the reaction media for the process 1) and process 2) are an organic solvent and an aqueous solution, respectively, it has long been thought impossible to achieve a one-pot chemo-enzymatic process. The appearance of a new methodology enabled us to demonstrate a one-pot process because the oxazoline formation has become possible in water without using any protecting groups and the resulting oxazolines can be utilized as effective glycosyl donors for the subsequent enzymatic glycosylation without isolation.
The synthesis of chitoheptaose has been achieved by using chitopentaose oxazoline as glycosyl donor and chitobiose as glycosyl acceptor catalyzed by a mutant chitinase.44) A combined use of one-step preparable oxazoline derivatives and an enzyme with a low hydrolytic activity enables the specific synthesis of chitoheptaose.
3-3. Application to glycoprotein synthesis.
It is well known that the molecular weight of glycoproteins produced by Chinese hamster ovary (CHO) cells are not homogeneous due to the heterogeneity of the oligosaccharide part (Fig. 27). It has become a problem for commercializing glycoproteins such as hormones and antibodies as biomedicines from the viewpoint of international standards. An efficient method for synthesizing homogenous glycoproteins is essential for producing some therapeutic glycoproteins or elucidating the role of glycans of glycoproteins.
We focused on the transglycosylation activity of endo-β-N-acetylglucosaminidase as a tool for glycoprotein syntheses, since this enzyme can catalyze not only the hydrolysis reaction of N-glycan but also the transglycosylation reaction of the oligosaccharide onto various acceptors having an N-acetylglucosamine residue.
First, a disaccharide substrate of Manβ1-4 GlcNAc-oxaozline was designed and synthesized as a novel probe for detecting the transglycosylation activity of endoglycosidases. Endo-M (from Mucor hiemalis) and Endo-A (from Arthrobacter protophormiae) have been found to recognize this substrate and afforded transglycosylated products in good yields. This is the first example of a transglycosylation reaction catalyzed by endo-β-N-acetylglcucosaminidases using a transition state analogue substrate.45)
Next, we demonstrated a transglycosylation catalyzed by a mutant enzyme by using a sialo-complex type sugar oxazoline as glycosyl donor. In combination with a simple preparation method for the sialo-complex-type sugar oxazoline substrate from natural sialoglycopeptide in egg yolk, the technique permits efficient synthesis of sialoglycosylated bioactive peptides and sialylated bovine ribonuclease B in about 90% yield.46)
The discovery of the direct synthesis of sugar oxazolines have accelerated the progress of synthetic research on glycoproteins with a homogeneous glycan. For example, various N-glycans were transferred from the corresponding oxazolines to the GlcNAc residue on the intact anti-Her2 antibody with a mutant endoglycosidase, affording glycoengineered anti-Her2 antibodies (Fig. 28).47) The binding assay of the resulting antibodies with homogenous N-glycans with FcγRIIIa-V158 showed that the glycoform influenced the affinity for FcγRIIIa-V158.
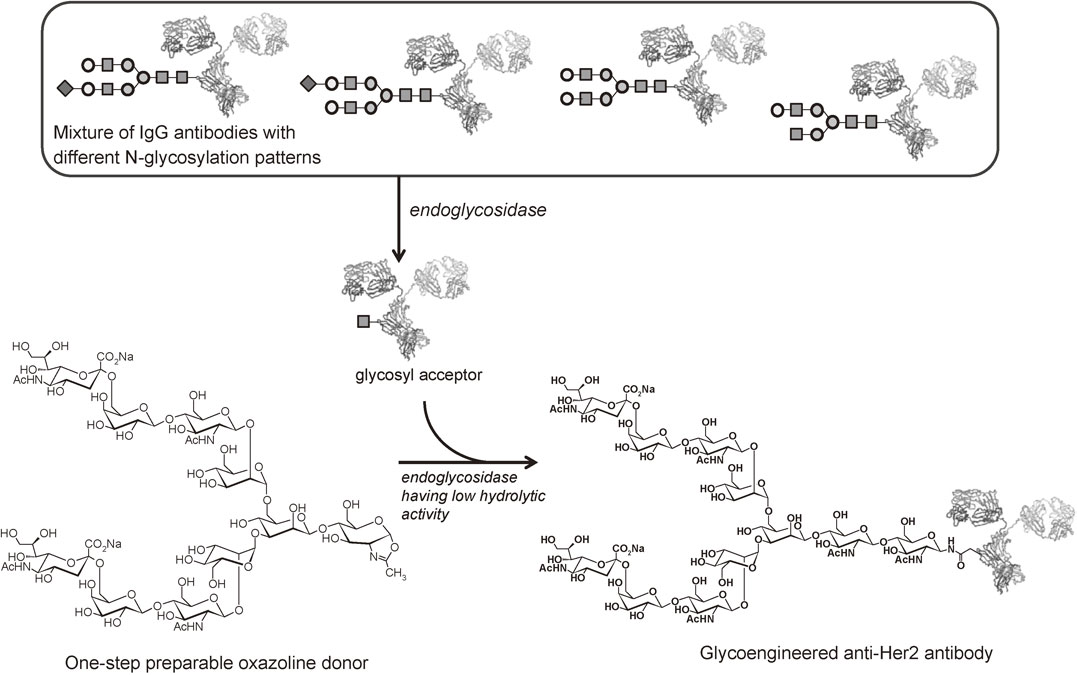
Davis et al. achieved the preparation of human IgG having a single glycoform from fucosylated IgG by remodeling using Endo-S (from Streptococcus pyogenes).48) Wang et al. developed an endoglycosidase having a lower hydrolysis activity from Endo-S by using the same strategy as that for the creation of Endo-M mutant. Endo-S whose Asp233 is replaced by Glu or Ala could catalyze transglycosylation reaction efficiently.49) Endo-S is a suitable enzyme for remodeling of IgG, since Endo-S recognizes IgG with high affinity. Endo-D (from Streptococcus pneumoniae) also recognized fucosylated N-linked oligosaccharides. The preparation of Endo-D having a decreased hydrolytic activity was achieved by the same strategy as Endo-M.50) The one-step preparation of sugar oxazolines is a general methodology that can be applied to various enzymatic transglycosylations in the glycotechnology field.
3-4. Protection-free synthesis of glycosyl compounds in aqueous media.
In general, carbohydrates are not soluble in organic solvents but soluble in water due to the good interactions between saccharides and water. On the other hand, almost all of the carbohydrates that exist in nature are glycosidic compounds as a result of dehydrative condensation of hemiacetal and aglycones. That means if one has to prepare glycosidic compounds in a flask, a dehydrative condensation reaction must be achievable in water. However, intermolecular dehydrative condensation is extremely disadvantageous in aqueous media due to the large negative values of standard Gibbs energy of formation for hydrolysis of glycosidic bonds.
We have recently proposed an efficient synthetic process for dehydrative glycosylation that consists of two elemental reactions, intramolecular dehydration and the subsequent addition reaction (Fig. 29). The first step is the enhancement of intramolecular dehydration which has an entropical favorability over intermolecular reactions. The second reaction is an intermolecular addition reaction where dehydration is unnecessary anymore because it has already been achieved in the first stage. If a nucleophile (ROH in Fig. 29) is suitably located in the vicinity of the reaction center, the addition reaction would be greatly driven by ring strain of the cyclic intermediate. The important thing is that a water molecule is removed not by a single reaction but through two elemental reactions, intramolecular dehydration and intermolecular addition. We focused on 1,2-anhydro sugars and sugar oxazolines as the cyclic synthetic intermediates having a ring strain (Fig. 30, center).

The first example is the one-step synthesis of 1,6-anhydro sugars 11 (Fig. 30).51) When an unprotected sugar is treated with 2-chloro-dimethylimidazolinium chloride (DMC) in water, the β-hydroxy group at C-1 reacts with DMC to give a reactive intermediate which is immediately converted to a 1,2-anhydo sugar. This step corresponds to the intramolecular dehydration in Fig. 29. The next addition reaction proceeds smoothly by the attack of a nucleophile, in this case, 6-hydroxy group, leading to the formation of 1,6-anhydro skeleton. The α-hydroxy group which is in equilibrium with the β-OH also reacts with DMC to afford an α-intermediate. However, this cannot be cyclized and immediately converted to the starting unprotected sugar with β-OH via an SN2 attack of water.
The 1,6-anhydro sugars are known to be useful synthetic intermediates for preparation of various glycosyl compounds such as S-glycosides, N-glycosides, glycosyl halides, C-glycosides, and proteoglycans. Further, 1,6-anydro sugars are important monomers for cationic ring-opening polymerizations, which afford stereo-regular polysaccharides. However, there have been no practical methods for their synthesis especially for 1,6-anhydro oligosaccharides with higher molecular weight in spite of their utility as precursors for functionalized polymers. The conventional method that uses pyrolysis of polysaccharides can only be applied to synthesis of 1,6-anhydro monosaccharides, because it is difficult to control the pyrolytic conditions to obtain a 1,6-anhydro oligosaccharide product selectively. The present method of using DMC in aqueous media would be a practical tool for synthesis of 1,6-ahnydro oligosaccharides.
(b) Protection-free synthesis of 1-thioglycoside derivatives.
Various 2-pyridyl 1-thioglycosides 12 and glycosyl dithiocarbamates 13, which are important synthetic intermediates and enzyme inhibitors in sugar chemistry, have been synthesized directly from the corresponding unprotected sugars by using DMC as a dehydrative condensing agent (Fig. 30).52),53) According to the present method, not only unprotected monosaccharides but also unprotected oligosaccharides such as cello-oligosaccharides, chito-oligo-saccharides, malto-oligosaccharides, and glucosamine oligomers, can be converted to the corresponding 2-pyridylthio derivatives, which would greatly expand the utility of aryl 1-thioglycosides in sugar chemistry.
(c) Protection-free synthesis of glycosyl azides.
Glycosyl azides are one of the most important synthetic intermediates in sugar chemistry. The recent developments of “click chemistry”54) have dramatically increased the potential of sugars possessing an azide function as precursors of glycoarrays and glycoconjugates.
Various oligosaccharides having an N-acetylglucosamine unit at the reducing end such as chitooligosaccharides and N-acetyllactosamine were converted to the corresponding β-glycosyl azides in good yields in the presence of 2,6-lutidine as a base.55) For the case of using strong bases like Et3N, sugar oxazoline derivatives are obtained as by-products. It is noteworthy that the present method could successfully be applied to disialo complex-type decasaccharide having two N-acetylneuramic acid moieties at the non-reducing ends, giving rise to the corresponding azide derivative without cleaving the inner glycosidic bonds.
Conclusions
Almost all naturally occurring carbohydrates exist as glycosidic compounds; they include oligo- or polysaccharides, glycolipids, glycoproteins, and nucleosides. In the biosynthesis of these compounds, the glycosidic bonds cannot be formed by the direct dehydration reactions of saccharide units and its aglycon parts. It is necessary to activate the anomeric center of the saccharide unit by introducing an appropriate leaving group so that the anomeric carbon atom is attacked by a hydroxy group of the aglycon part.
Catalysts responsible for the glycosylation of these activated saccharides are synthases, which are classified as glycosyltransferases. After being utilized, these glycosidic compounds are finally converted into carbon dioxide and water via combustion or degradation catalyzed by glycosidases from bacteria. It is, therefore, obvious that two kinds of enzymes, glycosyl transferases and glycosidases, are involved in the process of glycosylation and deglycosylation in nature, constructing a large carbon cycle system.
Based on the chemo-enzymatic glycosylation developed in our laboratory, a new carbon cyclic system which involves the production of functional glycosidic compounds such as artificial oligo- or polysaccharides has been proposed (“Glyco-chemistry Cycle System”) (Fig. 31).56),57) In this cyclic system, one glycosidase enzyme plays two roles including glycosidic bond formation and glycosidic bond cleavage by using the complementarity of glycosidases (see Fig. 13).

The chemo-enzymatic glycosylation concept consists of (1) transformation of a polysaccharide biomass to refined raw materials, (2) anomeric activation of the raw materials, (3) glycosylation catalyzed by a glycosidase to give functionalized oligosaccharides, (4) the degradation of the products by the glycosidase. This is a low environment-load process because the characteristic feature of glycosidases is to catalyze both glycosylation reaction and deglycosylation reactions due to the complementarity of glycosidases and, therefore, the resulting products can be converted to the starting materials enzymatically without using any drastic reaction conditions or strong acid catalysts.
All carbohydrate materials prepared by enzymes from natural origins can have a biodegradability that is tailored by the designer even if they have an artificial structure. The enzymatic glycosylation as well as the conventional chemical glycosylation will synergistically contribute not only to the creation of new glycotechnology, but also to the progress of the Green and Sustainable Chemistry.
For more than forty years, my research has strongly been concerned with the development of basic and general methodologies for glycoside synthesis. In particular, I have been focusing on the protection-free glycosylating process in these years, incorporating the new concept of “Direct Anomeric Activation” and “Glyco-Process Chemistry”. Neither the gorgeous total synthesis of complicated compounds nor the invention of intelligent glyco-materials have been my main interest. I have just been trying to find new reactions that can support a trunk of the tree of glyco-science by making full use of “Organic Synthesis” as the nutrients. Let me close by introducing a sentence from the writing of Georges Auguste Darzens (1867–1954) of the École polytechnique, showing the philosophy of science from a generalizer.58)
Les méthodes générales sont pour les chimistes ce que les formules sont pour les mathématiciens
(Generalized synthetic methods are to the chemist as theorems are to the mathematician)
Profile

Shin-ichiro Shoda was born in Tokyo, Japan in 1953. He graduated from the University of Tokyo, Department of Chemistry in 1976, and received his Ph.D. degree from the University of Tokyo in 1981 under the supervision of Professor Teruaki Mukaiyama in the field of synthetic organic chemistry, where he developed the glycosyl fluoride method as a novel glycosylating technology. After spending three years working as an Assistant Professor at the University of Tokyo, he conducted his postdoctoral fellowship at ETH-Zurich (from 1984 till 1986) with Professor Dieter Seebach. In 1986, he moved to Tohoku University and joined the lab of Professor Shiro Kobayashi in the field of polymer synthesis, where he developed new chemo-enzymatic glycosylations by using glycosyl fluorides and sugar oxazolines as glycosyl donors. In 1999, he was promoted to Full Professor at Tohoku University (Functional Macromolecular Chemistry Laboratory).
His research interests include synthesis of carbohydrates, development of novel glycosylations, macromolecular architecture and precision synthesis of well-designed functional oligo- and polysaccharides. He has been a Member of the Research Center for Science System at the Japan Society for the Promotion of Science (from 2003 till 2005). He received the Award of the Chemical Society of Japan for Young Chemists (1986), Science and Technology Institute Award for New Invention (1993), the Cellulose Society of Japan Award (2002), and Synthetic Organic Chemistry Award, Japan (2013).
Acknowledgements
The author would like to express his hearty thanks to Prof. Tamio Yamakawa, Professor Emeritus of the University of Tokyo and a Member of the Japan Academy for giving me this opportunity to provide this review. The author is indebted to Prof. Kenji Mori, Professor Emeritus of the University of Tokyo and a Member of the Japan Academy for many valuable suggestions that improved the manuscript. The author would like to express deep appreciation to Prof. Teruaki Mukaiyama and Prof. Shiro Kobayashi for providing me the chance to work in the synthetic field of carbohydrate chemistry. I would like to thank all the present and past collaborators including Prof. Jun-ichi Kadokawa, Dr. Masaya Fujita, Dr. Atsushi Kobayashi, Dr. Masato Noguchi, Dr. Gefei Li, Dr. Ryukou Izumi, Dr. Tomonari Tanaka, Dr. Tomohiko Kotake, Dr. Masaki Ishihara, Dr. Michinari Kohri, Dr. Wei Chun Huang, Dr. Naoki Yoshida, Dr. Kazunari Serizawa for their contribution to the development of new glycosylation reactions described herein. Appreciation is also extended to Prof. Takeshi Watanabe, Prof. Yasushi Morikawa, Prof. Tamou Fukamizo, Prof. Kenji Yamamoto, Prof. Kaoru Takegawa, Prof. Toshiyuki Inazu, Prof. Shinya Fushinobu, Prof. Takane Katayama, Dr. Motomitsu Kitaoka, Dr. Takashi Shirai for helpful discussion. Our work was supported by a Grant-in-Aid for Scientific Research from the Ministry of Education, Sports, Science and Technology, Industrial Technology Research Grant Program in 2006 from NEDO of Japan, and the International Center of Research & Education for Molecular Complex Chemistry in 2007 from Tohoku University Global COE Program. We also express our appreciation to Yaizu Suisankagaku Industry, Otsuka Chemical, Tokyo Kasei Kogyo, Fushimi Pharmaceutical for providing us with the oligosaccharides.
References
- 1) Shoda, S. (2013) Organic synthesis as culture. J. Synth. Org. Chem. Jpn. 71, 1115.
- 2) Shoda, S. (2013) Toward perfect glycosylation. Chemistry & Chemical Industry 66, 997–999.
- 3) Shoda, S., Kobayashi, A. and Kobayashi, S. (2016) Production of polymers by white biotechnology. In White Biotechnology for Sustainable Chemistry (eds. Coelho, M.A.Z. and Ribeiro, B.D.). Royal Society of Chemistry, Cambridge, pp. 274–309.
- 4) Koenigs, W. and Knorr, E. (1901) Ueber einige Derivate des Traubenzuckers und der Galactose. Berichte 34, 957–981.
- 5) Shoda, S. and Mukaiyama, T. (1979) A new method for the synthesis of β-glucosides using 2-chloro-3,5-dinitropyridine. Chem. Lett. 847–848.
- 6) Pougny, J.R., Jacquinet, J.C., Nassr, M., Duchet, D., Milat, M.L. and Sinaÿ, P. (1977) A novel synthesis of 1,2-cis-disaccharides. J. Am. Chem. Soc. 99, 6762–6763.
- 7) Schmidt, R.R. and Michel, J. (1980) Facile synthesis of α- and β-O-glycosyl imidates; preparation of glycosides and disaccharides. Angew. Chem. Int. Ed. Engl. 19, 731–732.
- 8) Shoda, S. (2008) Glycosyl fluorides. In Handbook of Chemical Glycosylation (ed. Demchenko, A.V.). Wiley-VCH, Weinheim, pp. 29–59.
- 9) Mukaiyama, T., Murai, Y. and Shoda, S. (1981) An efficient method for glucosylation of hydroxy compounds using glucosyl fluoride. Chem. Lett. 431–432.
- 10) Mukaiyama, T., Hashimoto, Y. and Shoda, S. (1983) Stereoselctive synthesis of 1,2-cis-glycofuranosides using glycofuranosyl fluorides. Chem. Lett. 935–938.
- 11) Hashimoto, S., Hayashi, M. and Noyori, R. (1984) Glycosylation using glucopyranosyl fluorides and silicon-based catalysits. Solvent dependency of the stereoselection. Tetrahedron Lett. 25, 1379–1382.
- 12) Nicolaou, K.C., Chucholowski, A., Dolle, R.E. and Randall, J.I. (1984) Reaction of glycosyl fluorides. Synthesis of O-, S-, and N-glycosides. Chem. Commun. 1155–1156.
- 13) Kreuzer, M. and Thiem, J. (1986) Aufbau von Oligosacchariden mit Glycosylfluoriden Unter Lewissaeure-Katalyse. Carbohydr. Res. 149, 347–361.
- 14) Matsumoto, T., Maeta, H., Suzuki, K. and Tsuchihashi, G. (1988) New glycosidation reaction 1. Combinational use of Cp2ZrCl2-AgClO4 for activation of glycosyl fluorides. Tetrahedron Lett. 29, 3567–3570.
- 15) Kanie, O., Ito, Y. and Ogawa, T. (1994) Orthogonal glycosylation strategy in oligosaccharide synthesis. J. Am. Chem. Soc. 116, 12073–12074.
- 16) Mootoo, D.R., Konradsson, P., Udodong, U. and Fraser-Reid, B. (1988) “Armed” and “Disarmed” n-pentenyl glycosides in saccharide couplings leading to oligosaccharides. J. Am. Chem. Soc. 110, 5583–5584.
- 17) Mukaiyama, T., Takeuchi, K., Jona, H., Maeshima, H. and Saitoh, T. (2000) A catalytic and stereoselective glycosylation with β-glycosyl fluorides. Helv. Chim. Acta 83, 1901–1918.
- 18) Nicolaou, K.C., Dolle, R.E., Papahatjis, D.P. and Randall, J.I. (1984) Practical synthesis of oligosaccharides. Partial synthesis of Avermectin B1a. J. Am. Chem. Soc. 106, 4189–4192.
- 19) Nicolaou, K.C., Caulfield, T.J., Kataoka, H. and Stylianides, N.A. (1990) Total synthesis of the tumor-associated Lex family of glycosphingolipids. J. Am. Chem. Soc. 112, 3693–3695.
- 20) Lee, Y.J., Lee, B.Y., Jeon, H.B. and Kim, K.S. (2006) Total synthesis of Agelagalastatin. Org. Lett. 8, 3971–3974.
- 21) Cumpstey, I., Fairbanks, A.J. and Redgrave, A.J. (2001) Sterospecific synthesis of 1,2-cis glycosides by allyl-mediated intramolecular aglycon delivery. 2. The use of glycosyl fluoride. Org. Lett. 3, 2371–2374.
- 22) Mori, K. and Tashiro, T. (2011) Sphingolipids and glycosphingolipids — their synthesis and bioactivities. Heterocycles 83, 951–1003.
- 23) Fischer, E. (1893) Ueber die Glucoside der Alkohole. Berichte 26, 2400–2412.
- 24) Ishihara, M., Takagi, Y., Li, G., Noguchi, M. and Shoda, S. (2013) Protection-free synthesis of alkyl glycosides under hydrogenolytic conditions. Chem. Lett. 42, 1235–1237.
- 25) Pasteur, L. (1897) Studies on fermentation. Macmillan Publishers.
- 26) Buechner, E. (1897) Alkoholische Gaehrung ohne Hefezellen. Berichte 30, 117–124.
- 27) Schmidt, R.R. (1986) New method for the synthesis of glycosides and oligosaccharide — Are there alternatives to the Koenigs-Knorr method? Angew. Chem. Int. Ed. Engl. 25, 212–235.
- 28) Kobayashi, S., Kashiwa, K., Kawasaki, T. and Shoda, S. (1991) Novel method for polysaccharide synthesis using an enzyme: the first in vitro synthesis of cellulose via a nonbiosynthetic path utilizing cellulase as catalyst. J. Am. Chem. Soc. 113, 3079–3084.
- 29) Shoda, S., Kawasaki, T., Obata, K. and Kobayashi, S. (1993) A facile enzymatic synthesis of cellooligosaccharide derivatives using β-lactosyl fluoride. Carbohydr. Res. 249, 127–137.
- 30) Shoda, S., Shintate, K., Ishihara, M. and Kobayashi, A. (2007) Colorimetric assay for evaluating glycosyl fluoride-hydrolyzing activity of glycosidase by using Allizarin Complexon reagent. Chem. Lett. 36, 16–17.
- 31) Kobayashi, S., Kiyosada, T. and Shoda, S. (1996) Synthesis of artificial chitin: irreversible catalytic behavior of a glycosyl hydrolase through a transition state analogue substrate. J. Am. Chem. Soc. 118, 13113–13114.
- 32) Kiso, M. and Anderson, L. (1979) The ferric chloride-catalyzed glycosylation of alcohols by 2-acylamido-2-deoxy-β-d-glucopyranose 1-acetates. Carbohydr. Res. 72, C12–C14.
- 33) Corey, E.J. (1988) Retrosynthetic thinking —essentials and examples—. Chem. Soc. Rev. 17, 111–133.
- 34) Tews, I., van Scheltinga, A.C.T., Perrakis, A., Wilson, K.S. and Dijkstra, B.W. (1997) Substrate-assisted catalysis unifies two families of chitinolytic enzymes. J. Am. Chem. Soc. 119, 7954–7959.
- 35) Shoda, S., Fujita, M., Lohavisavapanichi, C., Misawa, Y., Ushizaki, K., Tawata, Y., Kuriyama, M., Kohri, M., Kuwata, H. and Watanabe, T. (2002) Efficient method for the elongation of the N-acetylglucosamine unit by combined use of chitinase and β-galactosidase. Helv. Chim. Acta 85, 3919–3936.
- 36) Kohri, M., Kobayashi, A., Noguchi, M., Kawaida, S., Watanabe, T. and Shoda, S. (2006) Stepwise synthesis of chitooligosaccharides through a transition-state analogue substrate catalyzed by mutants of chitinase A1 from Bacillus circulanas WL-12. Holzforschung 60, 485–491.
- 37) Tanaka, T. (1978) Collapse of gels and the critical endpoint. Phys. Rev. Lett. 40, 820–823.
- 38) Raku, T. and Tokiwa, Y. (2001) Synthesis of thermosensitive polymers containing sugar branches. J. Appl. Polym. Sci. 80, 384–387.
- 39) Tanaka, T., Noguchi, M., Kobayashi, A. and Shoda, S. (2008) A novel glycosyl donor for chemo-enzymatic oligosaccharide synthesis: 4,6-dimethoxy-1,3,5-triazin-2-yl glycoside. Chem. Commun. 2016–2018.
- 40) Tanaka, T., Noguchi, M., Watanabe, K., Misawa, T., Ishihara, M., Kobayashi, A. and Shoda, S. (2010) Novel dialkoxytriazine-type glycosyl donors for cellulase-catalyzed lactosylation. Org. Biomol. Chem. 8, 5126–5132.
- 41) Noguchi, M., Nakamura, M., Ohno, A., Tanaka, T., Kobayashi, A., Ishihara, M., Fujita, M., Tsuchida, A., Mizuno, M. and Shoda, S. (2012) A dimethoxytriazine type glycosyl donor enables a facile chemo-enzymatic route toward α-linked N-acetylglucosaminyl-galactose disaccharide unit from gastric mucin. Chem. Commun. 48, 5560–5562.
- 42) Noguchi, M., Tanaka, T., Gyakushi, H., Kobayashi, A. and Shoda, S. (2009) Efficient synthesis of sugar oxazolines from unprotected N-acetyl-2-amino sugars by using chloroformamidinium reagent in water. J. Org. Chem. 74, 2210–2212.
- 43) Noguchi, M., Fujieda, T., Huang, W.C., Ishihara, M., Kobayashi, A. and Shoda, S. (2012) A practical one-step synthesis of 1,2-oxazoline derivatives from unprotected sugars and its application to chemoenzymatic β-N-acetylglucosaminidation of disialo-oligosaccharide. Helv. Chim. Acta 95, 1928–1936.
- 44) Yoshida, N., Tanaka, T., Noguchi, M., Kobayashi, A., Ishikura, K., Ikenuma, T., Seno, H., Watanabe, T., Kohri, M. and Shoda, S. (2012) One-pot chemoenzymatic route to chitoheptaose via specific transglycosylation of chitopentaose-oxazoline on chitinase-template. Chem. Lett. 41, 689–690.
- 45) Fujita, M., Shoda, S., Haneda, K., Inazu, T., Takegawa, K. and Yamamoto, K. (2001) A novel disaccharide substrate having 1,2-oxazoline moiety for detection of transglycosylating activity of endoglycosidases. Biochim. Biophys. Acta 1528, 9–14.
- 46) Umekawa, M., Higashiyama, T., Koga, Y., Tanaka, T., Noguchi, M., Kobayashi, A., Shoda, S., Huang, W., Wang, L.X., Ashida, H. and Yamamoto, K. (2010) Efficient transfer of sialo-oligosaccharide onto proteins by combined use of a glycosynthase-like mutant of Mucor hiemalis endoglycosidase and synthetic sialo-complex-type sugar oxazoline. Biochim. Biophys. Acta 1800, 1203–1209.
- 47) Kurogochi, M., Mori, M., Osumi, K., Tojino, M., Sugawara, S., Takashima, S., Hirose, Y., Tsukimura, W., Mizuno, M., Amano, J., Matsuda, A., Tomita, M., Takayanagi, A., Shoda, S. and Shirai, T. (2015) Glycoengineered monoclonal antibodies with homogeneous glycan (M3, G0, G2, and A2) using a chemoenzymatic approach have different affinities for FcγRIIIα and variable antibody-dependent cellular cytotoxicity activities. PLoS One 10, e0132848.
- 48) Goodfellow, J.J., Baruah, K., Yamamoto, K., Bonomelli, C., Krishna, B., Harvey, D.J., Crispin, M., Scanlan, C.N. and Davis, B.G. (2012) An endoglycosidase with alternative glycan specificity allows broadened glycoprotein remodelling. J. Am. Chem. Soc. 134, 8030–8033.
- 49) Huang, W., Giddens, J., Fan, S.Q., Toonstra, C. and Wang, L.X. (2012) Chemoenzymatic glycoengineering intact IgG antibodies for gain of functions. J. Am. Chem. Soc. 134, 12308–12318.
- 50) Fan, S.Q., Huang, W. and Wang, L.X. (2012) Remarkable transglycosylation activity of glycosynthase mutants of endo-D, an endo-β-N-acetylglucosaminidase from Streptococcus pneumoniae. J. Biol. Chem. 287, 11272–11281.
- 51) Tanaka, T., Huang, W.C., Noguchi, M., Kobayashi, A. and Shoda, S. (2009) Direct synthesis of 1,6-anhydro sugars from unprotected glycopyranoses by using 2-chloro-1,3-dimethylimidazolinium chloride. Tetrahedron Lett. 50, 2154–2157.
- 52) Yoshida, N., Noguchi, M., Tanaka, T., Matsumoto, T., Aida, N., Ishihara, M., Kobayashi, A. and Shoda, S. (2011) Direct dehydrative pyridylthio-glycosidation of unprotected sugars in aqueous media using 2-chloro-1,3-dimethylimidazolinium chloride as a condensing agent. Chem. Asian J. 6, 1876–1885.
- 53) Li, G., Noguchi, M., Kashiwagura, H., Tanaka, Y., Serizawa, K. and Shoda, S. (2016) Protection-free synthesis of glycosyl dithiocarbamates in aqueous media by using 2-chloroimidazolinium reagent. Tetrahedron Lett. 57, 3529–3531.
- 54) Kolb, H.C., Finn, M.G. and Sharpless, K.B. (2001) Click chemistry: diverse chemical function from a few good reactions. Angew. Chem. Int. Ed. Engl. 40, 2004–2021.
- 55) Tanaka, T., Nagai, H., Noguchi, M., Kobayashi, A. and Shoda, S. (2009) One-step conversion of unprotected sugars to β-glycosyl azides using 2-chloroimidazolinium salt in aqueous solution. Chem. Commun. 3378–3379.
- 56) Shoda, S. (2008) Glyco-chemistry cycle system based on glycosidases. In Experimental Glycoscience (eds. Taniguchi, N., Suzuki, A., Ito, Y., Narimatsu, H., Kawasaki, T. and Hase, S.). Springer, Tokyo, pp. 169–172.
- 57) Shoda, S., Izumi, R. and Fujita, M. (2003) Green process in glycotechnology. Bull. Chem. Soc. Jpn. 76, 1–13.
- 58) Laszlo, P. (1993) In Logique de la Synthèse Organique (Les Editions Ellipses).