Abstract
Cutibacterium acnes is a major commensal human skin bacteria. It is a producer of propionic acids that maintain skin acidic pH to inhibit the growth of pathogens. On the other hand, it is also associated with diseases such as acne vulgaris and sarcoidosis. C. acnes strains have been classified into six phylotypes using DNA-based approaches. Because several characteristic features of C. acnes vary according to the phylotype, the development of a practical method to identify these phylotypes is needed. For rapid identification of phylotypes for C. acnes strains, a matrix-assisted laser desorption/ionization mass spectrometry (MALDI-MS) fingerprinting technique has been applied; however, some phylotypes have not been discriminated. We developed a high-throughput protein purification method to detect biomarker proteins by ultrafiltration. MALDI-MS proteotyping using profiling of identified biomarker peaks was applied for the classification of 24 strains of C. acnes, and these were successfully classified into the correct phylotypes. This is a promising method that allows the discrimination of C. acnes phylotypes independent of a DNA-based approach.
Contributed by Koichi TANAKA, M.J.A.; Edited by Masanori OTSUKA, M.J.A.
Abbreviations: ACTH: adrenocorticotropic hormone; MALDI-MS: matrix-assisted laser desorption/ionization mass spectrometry; MDPNA: methylenediphosphonic acid; MLST: multi-locus sequence typing.
Introduction
Matrix-assisted laser desorption/ionization mass spectrometry (MALDI-MS) has been a powerful and approved tool for routine diagnostics in various fields such as the food industry and clinical microbiology. In conventional routine diagnostic methods, sample bacteria are analyzed by mass spectral fingerprinting, in which the sample bacterium is identified based on the similarity of mass spectra. It has already been evaluated for identification at the genus and species levels for a wide variety of clinically important bacteria and with reliable results.1),2) In contrast, bioinformatics-based approaches have been proposed for the rapid identification of bacteria.3),4) In this approach, detected peaks corresponding to bacterial proteins are assigned using calculated masses from amino acid sequences that are registered in public protein databases such as UniProt Knowledgebase (UniProtKB) and NCBI protein database. In addition, ribosomal subunit proteins (RSPs) have been proposed as ideal biomarker proteins because RSPs are one of the most abundant and typical housekeeping proteins that are practical and easily detected by MALDI-MS.5),6) However, intraspecies discrimination of bacteria is important for understanding the variety of bacterial-specific characteristics, such as virulence and drug resistance. Therefore, we developed a bacterial analysis method that discriminates bacteria at the subspecies and strain levels by MALDI-MS using RSPs as biomarkers.5),7)–11) In our previous study, the expressed RSPs from genome sequenced lactic acid bacteria strains of Lactobacillus plantarum,7) Lactobacillus delbrueckii subsp. bulgaricus, and Streptococcus thermophilus8) were examined using a bioinformatics-based approach. Although the amino acid sequences of RSPs are highly conserved, differences among same species can be detected using peak shifts, which reflect mutations in the amino acid sequences. These peak shifts are considered to be used as biomarkers for the classification of intraspecies bacterial strains. Furthermore, sixteen strains of Pseudomonas putida including different biovars and 21 strains of Rhodococcus erythropolis were classified based on profiling using RSPs detected by MALDI-MS. These obtained classification results for P. putida and R. erythropolis strains were highly similar to those based on the DNA gyrase subunit B gene sequencing.9),10) In addition, we looked at seventeen strains of Bifidobacterium longum, including three subspecies, which were successfully classified at the subspecies level.11) In our previous studies, we have not established a name for our bacterial classification method. In this study, our proposed bacterial classification method, which uses profiling of identified biomarker peaks is designated “MALDI-MS proteotyping”. This MALDI-MS proteotyping is similar to molecular typing method, multilocus sequence typing (MLST), and is based on a bioinformatics approach. Detected peaks in the MALDI mass spectra were then assigned using theoretical masses that enabled precise annotation of RSPs mutants and, therefore, can discriminate closely related bacterial strains. MALDI-MS proteotyping in which bacteria are discriminated based on sequence mutations is different from conventional mass spectral fingerprinting techniques, which are basically optimized for the identification of bacterial genera and species. MALDI-MS proteotyping is an attractive method that allows the analysis of multiple proteins independently from DNA-based techniques. However, it is necessary to compare the results of MALDI-MS proteotyping with those of genetic approaches to verify the discrimination resolution of MALDI-MS proteotyping of bacterial strains.
C. acnes is a Gram-positive anaerobic bacterium that is a major member of human skin commensal bacteria.12),13) The members of C. acnes are classified into three types, I, II, and III, based on traditional phenotypic characteristics such as serotypes, cell-wall sugar contents, fatty acid profiles, and morphology.14)–17) Recently, these were assigned to novel subspecies as follows: type I as C. acnes subsp. acnes,18) type II as C. acnes subsp. defendens,19) and type III as C. acnes subsp. elongatum.20) C. acnes plays an important role in maintaining skin health as a producer of propionic acid, which maintains acidic skin pH to prevent the growth of pathogens.21) C. acnes is also known as an opportunistic pathogen associated with acne vulgaris,22) sarcoidosis,23) prostate cancer,24) and device-related infections such as prosthetic joint infection.25),26) Because specific C. acnes strains display differences in interacting with microbe-related human diseases, it seems to be important to understand the diversity of C. acnes at the subtype/ribotype (RT) level.27) C. acnes strains are classified into six main phylotypes, designated IA1, IA2, IB, IC, II, and III by MLST.16),28)–31) Furthermore, C. acnes strains were classified at the RT level based on their unique 16S rRNA sequences.27) MALDI-MS fingerprinting has been applied to identify C. acnes phylotypes using two to five unidentified peaks.17),20),32) Even though the MALDI-MS fingerprinting technique is a powerful approach for rapid phylotype identification, the phylotypes were identified based on incomplete evidence due to using unidentified two to five peaks that were detected in the range m/z 6900–7500 and their doubly charged ions ([M + 2H]2+), which were detected in the range of m/z 3450–3750. In addition, IA2 and IB have not been discriminated yet.17),20),32) In the present study, MALDI-MS proteotyping was applied for 24 culture collection strains of C. acnes including five phylotypes, IA1, IA2, IB, II, and III (IA1: n = 5, IA2: n = 4, IB: n = 4, II: n = 8, and III: n = 3). We developed a practical and reliable classification method for C. acnes strains at the phylotype level based on MALDI-MS proteotyping. In addition, classification of IC based on theoretical masses calculated from amino acid sequences on a protein database was also discussed.
Materials and methods
Bacterial strains.
The bacterial strains used in this study were provided by the RIKEN BRC (Tsukuba, Ibaraki, Japan) through the National BioResource Project of the MEXT/AMED, Japan. Table 1 summarizes the 24 sample strains of C. acnes with their phylotypes and MLST sequence types (STs).33) The sample strains were cultured in Gifu anaerobic medium (GAM) broth (Nissui Pharmaceutical Co., Tokyo, Japan) in anaerobic conditions at 37 °C for 48 to 72 hours.
Table 1. Sample strains of
C. acnes
| Culture collection number |
Phylotypea |
Sequence type |
Sample namea |
| C. acnes subsp. acnes (type I) |
| JCM |
6425T |
IA1 |
6b |
— |
| JCM |
6495 |
IA1 |
— |
— |
| JCM |
18916 |
IA1 |
6a |
K107 |
| JCM |
18922 |
IA1 |
6a |
K161 |
| JCM |
18924 |
IA1 |
6a |
K282 |
| JCM |
18907 |
IA2 |
22a |
K51 |
| JCM |
18908 |
IA2 |
9a |
K56 |
| JCM |
18910 |
IA2 |
22a |
K72 |
| JCM |
18912 |
IA2 |
9a |
K94 |
| JCM |
18917 |
IB |
10a |
K114 |
| JCM |
18918 |
IB |
10a |
K115 |
| JCM |
18923 |
IB |
10a |
K280 |
| JCM |
18927 |
IB |
10a |
S16 |
| C. acnes subsp. defendens (type II) |
| JCM |
6473T |
II |
— |
— |
| JCM |
18911 |
II |
43a |
K80 |
| JCM |
18913 |
II |
46a |
K96 |
| JCM |
18914 |
II |
43a |
K104 |
| JCM |
18915 |
II |
43a |
K106 |
| JCM |
18920 |
II |
43a |
K127 |
| JCM |
18921 |
II |
43a |
K145 |
| JCM |
18926 |
II |
42a |
S13 |
| C. acnes subsp. elongatum (type III) |
| JCM |
18909 |
III |
19a |
K57 |
| JCM |
18919T |
III |
19a |
K124 |
| JCM |
18925 |
III |
19a |
K290 |
a: Dekio et al. J Med Microbiol (2012) 61, 622–630.
b: McDowell et al. Microbiology (2011) 157, 1990–2003.
Bacterial samples were prepared at three different protein purification levels to optimize sample preparation for detecting RSPs: whole cell for the direct smear method, cell lysate for the premixed method, and protein fraction for the premixed method. Each of the 24 C. acnes strains were harvested by centrifugation and washed in Milli-Q water (Millipore Co., Bedford, MA, USA). The whole cell sample were bacterial cells suspended in Milli-Q water (OD600 = 1). Preparation of cell lysates was carried out based on the methods reported previously.8)–10) Briefly, bacterial cells were ground with zirconia beads using a Micro Smash™ MS-100 (Tomy Seiko Co. Ltd., Tokyo, Japan) at 5000 rpm three times of 60 s each, and beads and cell debris were removed by centrifugation (15000 g, 5 min). Protein fractions were obtained by ultra-filtration of the cell lysates (14000 g, 10 min). Centrifugal filter devices (Amicon Ultra™, NMWL 10 kDa, Millipore Co.) were used to capture ribosome particles. The scheme is illustrated in Fig. 1.

The theoretical masses of RSPs were calculated for the following genome sequenced strains used in this study, C. acnes subsp. acnes JCM 6425T, JCM 18916, and JCM 18918, C. acnes subsp. defendens JCM 6473T and JCM 18920, C. acnes subsp. elongatum JCM18909. Amino acid sequences of each RSP were obtained from the NCBI protein database (https://www.ncbi.nlm.nih.gov/protein/). The calculated mass of each RSP was predicted using the Compute pI/Mw tool on the ExPASy proteomics server (https://web.expasy.org/compute_pi_tool.html), with N-terminal methionine loss considered first as a possible post-translational modification. The possibilities of other modifications will be discussed below in the Results and Discussion sections. The theoretical mass of each ribosomal protein was calculated as [M + H]+.
MALDI-MS.
Sinapinic acid (SA) matrix solution at a concentration of 10 mg/mL in 50% acetonitrile (ACN) with 1% trifluoroacetic acid (TFA) was used for MALDI-MS sample preparation. Optimization of sample preparation was carried out using a cell suspension of C. acnes JCM 6425T (OD600 = 1).
For the direct smear method, 1 µL of each cell suspension was spotted onto the MALDI target and dried in air. To extract RSPs from C. acnes strains, formic acid (FA) extraction, which was used for microorganisms such as yeasts and Corynebacterium, was performed.34),35) On-plate extraction using 0.5 µL of 30% FA was carried out before dropping of 1 µL of SA solution onto the sample. For preparation of the premixed method, 1 µL of each of cell lysate or protein fraction was mixed with 10 µL of SA matrix solution. One µL of sample/matrix mixture was spotted onto the MALDI target and dried in air. Methylenediphosphonic acid (MDPNA)36) solution at a concentration of 1% (w/v) was prepared with 50% ACN with 1% TFA solution. The MDPNA solution was used for the preparation of a matrix solution with MDPNA. More than 7 sample spots were prepared for each samples.
The MALDI-MS measurements were performed using an AXIMA Performance™ (Shimadzu/Kratos, Manchester, UK) mass spectrometer equipped with a pulsed nitrogen UV laser (337 nm) in the positive ion linear mode. More than seven MALDI mass spectra for each sample were acquired from independent sample spots in the range of m/z 2000–30000. External mass calibration was carried out using two peaks of adrenocorticotropic hormone (ACTH) 18–39 ([M + H]+, m/z 2466.7) and myoglobin ([M + H]+, m/z 16952.6) followed by self-calibration using moderately strong peaks assigned to RSPs as internal references. The peak matching of the biomarker proteins was judged from tolerance within 200 ppm. Peak matching and statistical analysis were carried out using eMSTAT Solution™ software (Shimadzu Corp., Kyoto, Japan).
Identification of biomarker protein.
Unidentified biomarker protein detected at m/z 7035 was purified from 250 µL of the filtration fraction in Fig. 1. It was purified using a step gradient elution with 10%, 20%, and 30% ACN solution using Bond Elut Omix C4 100 µL pipette tips (Agilent Technologies Inc., Santa Clara, CA, U.S.A.). The unidentified biomarker protein was eventually eluted in the 30% ACN fraction. The obtained fraction was evaporated and dissolved in 25 µL of Milli-Q water. Tryptic peptides from the unidentified protein were obtained using sequencing grade modified trypsin (Promega, Madison, WI, U.S.A.) with incubation at 37 °C for 8 hours. After demineralization using ZipTip C4 (Millipore, Bedford, MA, U.S.A.), the digested sample was mixed with α-cyano-4-hydroxy cinnamic acid at a concentration of 10 mg/mL in 50% ACN solution containing 1% TFA and subjected to MALDI-MS measurement. Identification of the biomarker protein detected at m/z 7035 was carried out by using Mascot peptide mass fingerprinting (PMF) search against the NCBInr database using MASCOT (version 2.6.1, Matrix Science Ltd., London, U.K.). The database search parameters were as follows: taxonomic setting was other Actinobacteria, the number of allowed missed cleavage was 2, and average mass tolerance was set at ±150 ppm. Identification was judged if a significance score p < 0.05 was obtained.
Cluster analysis.
The matching of observed masses to the calculated masses was judged from a tolerance within 200 ppm. The judgement results for each biomarker protein were summarized in the binary biomarker matching table, in which scores used either 1 or 0. A dendrogram was constructed using the unweighted pair group method with arithmetic mean (UPGMA) obtained from the Euclidian distance.
Results and discussion
Detection of RSPs from type strain of C. acnes subsp. acnes.
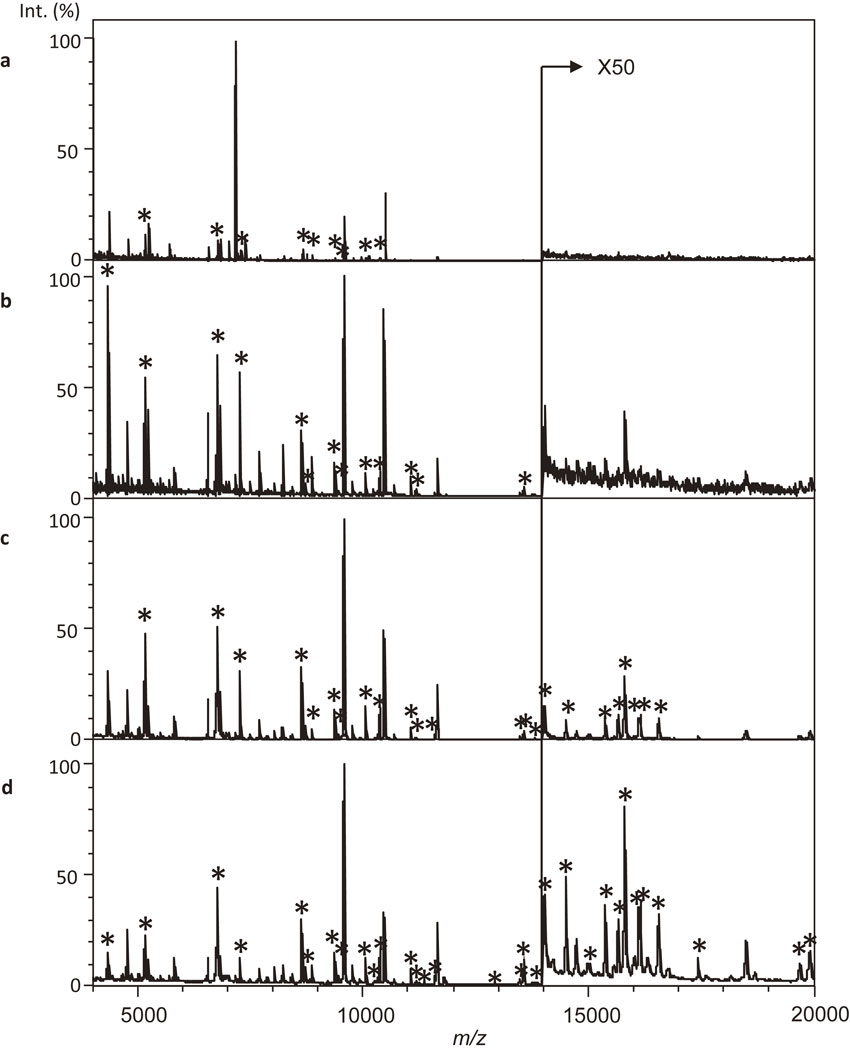
In this study, we examined bacterial sample preparation methods to detect RSPs, which are promising biomarkers to bacterial phylogenetic classification with C. acnes subsp. acnes JCM 6425T. Figure 2 shows a comparison of MALDI-MS data from bacterial samples from whole cells (a), cell lysate (b), protein fraction (c), and protein fraction with MDPNA (d); asterisks (*) indicate RSPs. The number of detected RSPs in each MALDI-MS was as followings, whole cell, 9; cell lysate, 13; protein fraction, 22; and protein fraction with MDPNA, 31. These results suggested that sample preparation using a protein fraction with MDPMA was sufficient to detect significant numbers of RSPs. A total of nine peaks corresponding to RSPs were detected from the whole cell sample prepared using direct smear method with FA extraction (Fig. 2a). Although the peaks corresponding to RSPs were relatively low, two peaks used for MALDI-MS fingerprinting17),20),32) were detected at m/z 7035 and m/z 7180 with higher intensities. Because C. acnes is not only a member of Gram-positive bacteria but also a biofilm producer,37)–39) it seemed to be difficult to extract sufficient amounts of RSPs using cell lysis with an organic solvent and organic acids. In addition, microbial constituents should have negative effects on the ionization of RSPs. Cell disruption by bead beating and removing debris by centrifugation was effective to detect RSPs; however, RSPs were not observed in the range larger than m/z 14000 (Fig. 2b). Although it is easy to detect RSPs from purified proteins by MALDI-MS, treatment by ultracentrifugation was needed for at least 1 hour.7)–10),40),41) Here, the effect of ultracentrifugation on the detection of RSPs was to concentrate the RSPs and remove low-molecular-weight components such as minerals and peptides. We examined the purification of RSPs using ultrafiltration instead of ultracentrifugation to develop a purification method for RSPs with commonly available equipment and in a time-effectively manner. Purification of the protein fraction by ultrafiltration took only 5–10 minutes with a commonly used centrifuge with cooling function (14000 g at 4 °C). Figure 2c shows MALDI-MS data of protein fraction prepared by ultrafiltration. RSPs were detected in the range of m/z 4000–20000 by ultrafiltration. RSPs exist in bacterial cells as ribosome particles, and these are easily captured by ultrafiltration. Although RSPs were not completely purified by ultrafiltration, this method seemed to be sufficient to detect RSPs by MALDI-MS. Moreover, adding MDPNA improved the detection of RSPs in the higher mass range (Fig. 2d). MDPNA has been proposed as additive to analyze samples that contain LC buffer. The effect of MDPNA was described as follows, improved S/N ratio, reduced generation of unnecessary adduct ions, reduced salt-induced signal suppression, and pH adjustment. The reducing salt-induced signal suppression and acidification by MDPNA was considered to be effective for clear detection of RSPs. Therefore, protein fractions prepared with MDPNA appeared to be sufficient for the analysis of RSPs of C. acnes strains. Details of the observed peaks are supplied in supplementary materials Table S1. We identified a number of errors in the amino acid sequences of RSPs registered in the public protein databases based on comparisons with the observed masses. The errors in amino acid sequences of registered proteins were corrected by comparison with registered amino acid sequences of other closely related genome sequenced strains. Corrected amino acid sequences for the assignment of detected peaks have been summarized with their related information such as accession numbers (supplementary materials Table S1-2 and S1-3).
Peak assignment for the detected peaks from each genome-sequenced strain, C. acnes subsp. acnes JCM 6425T (IA1), JCM 18916 (IA1), and JCM 18918 (IB), C. acnes subsp. defendens JCM 6473T (II) and JCM 18920 (II), and C. acnes subsp. elongatum JCM 18909 (III), was carried out based on comparison with the theoretical masses calculated from the amino acid sequences of each RSP. The number of detected RSPs differed among the genome-sequenced strains. In addition to N-terminal methionine loss, which is known as a major post-translational modification for RSPs,6) other post-translational modifications such as disulfide binding, methylation, and acetylation are also detected in bacterial RSPs. For example, disulfide bonding, which was predicted based on a C–x–x–C motifs (C: cysteine, x: any amino acid residue) are detected in L36, L28, L31, L33, and S14. Even though disulfide binding is easy to predict from amino acid sequences, these RSPs with a C–x–x–C motif are removed from biomarkers because peak shifts by −4 or −2 were not always detected in this study. Furthermore, RSPs whose mass differences were smaller than 200 ppm, such as L7/12 (m/z 13571.4) and L18 (m/z 13570.6), were removed from the biomarkers because these peaks were too close to discriminate from each other. About 25 to 30 RSPs were assigned for all the sample strains with repeatability in the range of m/z 4000–20000 (Table SI-2 and SI-3.). A total of 11 RSPs summarized in Table 2 are biomarker candidates to classify C. acnes phylotypes. These RSPs were commonly detected from six genome-sequenced strains used in this study. Each of the six biomarker proteins, L9, L29, L30, S8, S15, and S19, were detected at the same m/z values among the genome-sequenced strains used in this study. In contrast, there were slight mutations in amino acid sequences in L6, L13, L15, and L23, and these RSPs were expected to contribute to the classification of C. acnes strains.
Table 2. Biomarker candidates selected from RSPs
| Protein name |
Calculated masses |
| L6 |
1) 19678.6 |
2) 19706.6 |
|
| L9 |
1) 16118.8 |
|
|
| L13 |
1) 16153.5 |
2) 16167.6 |
3) 16181.6 |
| L15 |
1) 15357.6 |
2) 15384.7 |
|
| L23 |
1) 11181.0 |
2) 11200.0 |
|
| L27 |
1) 9362.4 |
2) 9392.4 |
|
| L29 |
1) 8754.9 |
|
|
| L30 |
1) 6786.9 |
|
|
| S8 |
1) 14525.7 |
|
|
| S15 |
1) 10080.7 |
|
|
| S19 |
1) 10380.0 |
|
|
The biomarker candidates to classify C. acnes phylotypes in Table 2 were reproducibly detected from all of the sample strains used in this study. Mass-matching profiles of the RSP biomarkers, which were denoted as 0 or 1 (1: detected, 0: not detected) and the resultant dendrogram together with their types and phylotypes is shown in Fig. SI-1. Types I, II, and III were classified using RSP biomarkers. Here, the most important issue in this study was differentiation of each phylotype within C. acnes subsp. acnes (type I), IA1, IA2, and IB. From this perspective, L13 was a crucially important biomarker protein to discriminate IB from IA1 and IA2, which was impossible to discriminate using the fingerprinting method.17) Although IB was differentiated from IA1 and IA2, IA1 and IA2 were not discriminated. Furthermore, C. acnes subsp. acnes JCM 18916 (IA1) deviated from the IA1 cluster because of the mutation of amino acid residue in L27. At present (July 26, 2019), more than 250 amino acid sequences of L27 for C. acnes are registered in the protein databases. Here, the amino acid sequence of L27 of C. acnes JCM 18916 is supposed to be unique in C. acnes strains, because only 5 strains (HL002PA2, HL002PA3, HL036PA2, HL036PA3, and HL046PA2) had the same sequences. Although L27 seemed to be a potentially effective biomarker protein to further classify IA1 strains, this RSP was removed from biomarker protein in order to make a single cluster for IA1 and also to classify C. acnes subsp. acnes JCM 18916 into the IA1 cluster. In addition, strains of type II were classified into two clusters based on L13, whose sequence types (STs) analyzed by MLST were 42 and 46, and others.42) In order to discriminate IA1 and IA2, a biomarker candidate was selected using statistical analysis with eMSTAT Solution software. As a result, it was indicated that a peak detected at m/z 7035 was specific for IA1 strains. Although this peak had not been characterized, it has been used by other researchers17),20),32) to discriminate phylotypes of C. acnes strains using the MALDI-MS fingerprinting approach. Because the peak was detected with high intensity using the direct smear method using the whole cell (Fig. 2a), it was considered to be a non-cytoplasmic protein. In addition, the protein was presumed to be a non-ribosomal protein because it was removed from cell-lysates by ultrafiltration. Furthermore, proteome of C. acnes ATCC 6919T (= JCM 6425T) has already been studied.43) In the study, CsbD-like protein and hypothetical protein PPA1018 were reported as abundant cell wall proteins whose molecular weight was about 7000. First, we calculated the theoretical masses of these proteins. The calculated m/z values for the [M + H]+ ion of CsbD-like protein was 7179.9, and that for hypothetical protein PPA1018 was 7034.6. Moreover, hypothetical protein PPA1018 was identified as an antitoxin by homology searches for amino acid sequences of the protein. Amino acid sequences and calculated masses considering N-terminal methionine loss are summarized with their accession numbers in Table 3. To verify our prediction, we also analyzed whole cells of sample strains using the direct smear method. MALDI-MS data from each phylotype of genome-sequenced strains in the range m/z 6900–7300 are shown in Fig. 3. Two to three peaks were detected at different m/z values reflecting their phylotypes. Peaks were successfully matched with the calculated masses for antitoxins and CsbD-like proteins. Our data supported the results reported by Dekio et al.,17),20) and these results suggested that IA2 and IB were not distinguished using antitoxin and CsbD-like protein as biomarkers. On the other hand, Nagy et al. reported that IA1 and IA2 were not distinguished by MALDI-MS fingerprinting technique, because both of their characteristic peaks were in common at m/z 7034 and 7180.32) Although their results did not agree with ours, the theoretical masses of antitoxin for three strains of C. acnes phylotype IA2 (P.acn17, P.acn31, and P.acn33) used in their study32) were m/z 7005, which agreed with our assumption. Moreover, the predominant peaks detected in the range of m/z 3450–3650 and m/z 6950–7300 were reported.20) These peaks seemed to be [M + 2H]2+ and [M + H]+ corresponding to antitoxin and CsbD-like protein for each phylotype. Here, we decided to add antitoxin, which was detected at m/z 7035 as biomarker proteins to discriminate IA1 and IA2. In addition, PMF analysis of purified antitoxin of C. acnes subsp. acnes JCM 6425T was made from fragments derived through trypsin digestion (Fig. SI-2a). The expected tryptic masses clearly matched the calculated values within 150 ppm differences, as shown in Fig. SI-2b. This result also supported our prediction that the peak detected at m/z 7035 corresponded to an antitoxin. We examined the effectiveness of these two kinds of proteins as biomarkers. Although antitoxin and CsbD-like protein were major peaks when bacterial samples were prepared using the direct smear method, these were not major peaks in the MALDI-MS data of protein fractions. In addition, antitoxin of C. acnes subsp. elongatum was not always detected as reported by Dekio et al.17),20) For these reasons, we decided that addition of antitoxin to IA1 as a biomarker was suitable. In other words, observing the presence or absence of antitoxin for IA1 was more suitable for the discrimination of IA1 and IA2. The biomarker matching table (Table 4) was processed by UPGMA cluster analysis to generate a dendrogram. Figure 4 shows the resulting dendrogram of C. acnes strains together with their phylotypes. A total of 24 C. acnes strains used in this study were split into three major clusters that correlated with traditional phylogenetic classification defined as types I, II, and III by two RSPs selected from L23, L15, and L6. In addition, members of type I were classified into phylotypes as IA1, IA2, and IB by L13 and antitoxin. As a result, we successfully classified 24 strains of C. acnes phylotypes based on 10 RSPs and antitoxin for IA1. Here, classification of IC could be evaluated on a theoretical level, because we did not have IC strains. Theoretical masses of the biomarkers for IC strains were able to be calculated from their amino acid sequences of genome sequenced IC strains, HL097PA1 and PRP-38, and are summarized in Table SI-4. Based on the estimation from amino acid sequences of each protein of IC strains, IC strains were considered to be discriminated from other phylotypes by L23. It should be noted that IC strains will not be discriminated from IA2 and IB by the fingerprinting method using two peaks, which were assigned as antitoxin and CsbD-like protein, because the calculated masses of these proteins were same as IA2 and IB (Table SI-4). This is the first report that has successfully classified IA1, IA2, and IB by MALDI-MS proteotyping. It is important to detect as many biomarkers as possible to better understand the diversity of C. acnes strains. On the other hand, we proposed a workflow for the rapid identification of phylotypes of C. acnes strains by MALDI-MS proteotyping in Fig. 5. This workflow enabled the identification of C. acnes phylotypes IA1, II, and III with a combination of two biomarker proteins, antitoxin and CsbD-like protein, and IA2, IB, and IC were identified with a combination of five biomarker proteins, antitoxin, CsbD-like protein, L6, L13, and L23. Our proposed workflow should be effective for the identification of phylotypes for a large number of C. acnes strains.
Table 3. Amino acid sequences of antitoxin and CsbD-like protein with calculated masses
| Antitoxin (66 aa) |
| Calculated mass: m/z 7034.6 (IA1) |
| MGLFDKAKDAISDRQDDIKNQASQHSDQVEQGIDKAGNTVDDKTGGKFSDQIDKGQDALKDKLGDL |
| Calculated mass: m/z 7004.5 (IA2, IB, IC, and III) |
| MGLFDKAKDAISDRQDDIKNQASQHSDQVEQGIDKAGNAVDDKTGGKFSDQIDKGQDALKDKLGDL |
| Calculated mass: m/z 6985.5 (II) |
| MGLFDKAKDAISDHQDDIKNQASQHSDQVEQGIDKAGNAVDDKTGGKFSDQIDKGQDALKDKLGDL |
| CsbD-like protein (71 aa) |
| Calculated mass: m/z 7179.9 (IA1, IA2, IB, IC and III) |
| MGLSDKINSKSDEAVGAAKEKIGGLTDDSDLKSAGADQKASGKVAQKVEDVKDKANDLKHNVQAAADKLKG |
| Calculated mass: m/z 7238.0 (III) |
| MGLSDKINSKSDEAVGAAKEKIGGLTDDSDLKSEGADQKASGKVAQKVEDVKDKANDLKHNVQAAADKLKG |
| Calculated mass: m/z 7251.0 (II, ST 42 and 46) |
| MGLSDKINSKSDEAVGAAKEKIGGLTDDSDLKSEGANQKASGKVAQKVEDVKDKANDLKHNIQAAADKLKG |
| Calculated mass: m/z 7265.0 (II, ST 43) |
| MGLSDKINSKSDEAVGAAKEKIGGLTDDSDLKSEGANQKASGKVAQKVEDVKDKANDLKHNVQAVADKLKG |
Underlines in the amino acid sequences indicate mutated amino acid residues.

Table 4. Biomarker matching table in a binary format of the sample strains
| Strains |
Biomarker proteins |
| Phylotype |
JCM |
L9 |
L29 |
L30 |
S8 |
S15 |
S19 |
Antitoxin |
L6 |
L13 |
L15 |
L23 |
| 16118.8 |
8754.9 |
6786.9 |
14525.7 |
10080.7 |
10380.0 |
7034.6 |
19678.6 |
19706.6 |
16153.5 |
16167.6 |
16181.6 |
15357.6 |
15384.7 |
11181.0 |
11200.0 |
| IA1 |
6425T |
1 |
1 |
1 |
1 |
1 |
1 |
1 |
0 |
1 |
0 |
1 |
0 |
0 |
1 |
0 |
1 |
| 6495 |
1 |
1 |
1 |
1 |
1 |
1 |
1 |
0 |
1 |
0 |
1 |
0 |
0 |
1 |
0 |
1 |
| 18916 |
1 |
1 |
1 |
1 |
1 |
1 |
1 |
0 |
1 |
0 |
1 |
0 |
0 |
1 |
0 |
1 |
| 18922 |
1 |
1 |
1 |
1 |
1 |
1 |
1 |
0 |
1 |
0 |
1 |
0 |
0 |
1 |
0 |
1 |
| 18924 |
1 |
1 |
1 |
1 |
1 |
1 |
1 |
0 |
1 |
0 |
1 |
0 |
0 |
1 |
0 |
1 |
| IA2 |
18907 |
1 |
1 |
1 |
1 |
1 |
1 |
0 |
0 |
1 |
0 |
1 |
0 |
0 |
1 |
0 |
1 |
| 18908 |
1 |
1 |
1 |
1 |
1 |
1 |
0 |
0 |
1 |
0 |
1 |
0 |
0 |
1 |
0 |
1 |
| 18910 |
1 |
1 |
1 |
1 |
1 |
1 |
0 |
0 |
1 |
0 |
1 |
0 |
0 |
1 |
0 |
1 |
| 18912 |
1 |
1 |
1 |
1 |
1 |
1 |
0 |
0 |
1 |
0 |
1 |
0 |
0 |
1 |
0 |
1 |
| IB |
18917 |
1 |
1 |
1 |
1 |
1 |
1 |
0 |
0 |
1 |
1 |
0 |
0 |
0 |
1 |
0 |
1 |
| 18918 |
1 |
1 |
1 |
1 |
1 |
1 |
0 |
0 |
1 |
1 |
0 |
0 |
0 |
1 |
0 |
1 |
| 18923 |
1 |
1 |
1 |
1 |
1 |
1 |
0 |
0 |
1 |
1 |
0 |
0 |
0 |
1 |
0 |
1 |
| 18927 |
1 |
1 |
1 |
1 |
1 |
1 |
0 |
0 |
1 |
1 |
0 |
0 |
0 |
1 |
0 |
1 |
| II |
6473T |
1 |
1 |
1 |
1 |
1 |
1 |
0 |
1 |
0 |
0 |
0 |
1 |
1 |
0 |
0 |
1 |
| 18911 |
1 |
1 |
1 |
1 |
1 |
1 |
0 |
1 |
0 |
0 |
0 |
1 |
1 |
0 |
0 |
1 |
| 18914 |
1 |
1 |
1 |
1 |
1 |
1 |
0 |
1 |
0 |
0 |
0 |
1 |
1 |
0 |
0 |
1 |
| 18915 |
1 |
1 |
1 |
1 |
1 |
1 |
0 |
1 |
0 |
0 |
0 |
1 |
1 |
0 |
0 |
1 |
| 18920 |
1 |
1 |
1 |
1 |
1 |
1 |
0 |
1 |
0 |
0 |
0 |
1 |
1 |
0 |
0 |
1 |
| 18921 |
1 |
1 |
1 |
1 |
1 |
1 |
0 |
1 |
0 |
0 |
0 |
1 |
1 |
0 |
0 |
1 |
| 18913 |
1 |
1 |
1 |
1 |
1 |
1 |
0 |
1 |
0 |
0 |
1 |
0 |
1 |
0 |
0 |
1 |
| 18926 |
1 |
1 |
1 |
1 |
1 |
1 |
0 |
1 |
0 |
0 |
1 |
0 |
1 |
0 |
0 |
1 |
| III |
18909 |
1 |
1 |
1 |
1 |
1 |
1 |
0 |
1 |
0 |
1 |
0 |
0 |
0 |
1 |
1 |
0 |
| 18919T |
1 |
1 |
1 |
1 |
1 |
1 |
0 |
1 |
0 |
1 |
0 |
0 |
0 |
1 |
1 |
0 |
| 18925 |
1 |
1 |
1 |
1 |
1 |
1 |
0 |
1 |
0 |
1 |
0 |
0 |
0 |
1 |
1 |
0 |

This is the first report that has successfully classified the phylotypes of C. acnes strains as IA1, IA2, IB, II, and III based on MALDI-MS proteotyping using assigned biomarker proteins such as RSPs and antitoxin. Although IC strains were not available for this study, theoretical masses, which were calculated from the amino acid sequences for IC strains, indicated that the phylotypes for IC strains will be identified correctly. Further study using IC strains is required to verify our approach. Although MLST is widely recognized as the gold-standard technique allowing the discrimination of phylotypes of C. acnes, our proposed method is still a viably important workflow, which enabled us to discriminate phylotypes of C. acnes independently from a DNA-based technique.
Supplementary materials
Table SI-1: Detected ribosomal proteins of C. acnes JCM 6425T. Table SI-2-1 to SI-2-24: Tables for detected ribosomal proteins of sample strains. Table SI-3: Corrected amino acid sequences of biomarker RSPs of genome sequenced strains. Table SI-4: Calculated masses for phylotype IC strains. Fig. SI-1: Biomarker matching table in a binary format of the sample strains (a) and resultant dendrogram of C. acnes strains based on variation of the 11 RSPs (b). Fig. SI-2: MALDI peptide mass fingerprint spectrum of the protein detected at m/z 7035 (a) and matched peptide fragments from the tryptic digestion of the protein (b). The sequence coverage of these fragments is shown in bold.
These materials are available free at https://doi.org/10.2183/pjab.95.042.
References
- 1) Dubois, D., Segonds, C., Prere, M.F., Marty, N. and Oswald, E. (2013) Identification of clinical Streptococcus pneumoniae isolates among other alpha and nonhemolytic streptococci by use of the Vitek MS matrix-assisted laser desorption ionization-time of flight mass spectrometry system. J. Clin. Microbiol. 51, 1861–1867.
- 2) Eigner, U., Holfelder, M., Oberdorfer, K., Betz-Wild, U., Bertsch, D. and Fahr, A.M. (2009) Performance of a matrix-assisted laser desorption ionization-time-of-flight mass spectrometry system for the identification of bacterial isolates in the clinical routine laboratory. Clin. Lab. 55, 289–296.
- 3) Demirev, P.A., Lin, J.S., Pineda, F.J. and Fenselaut, C. (2001) Bioinformatics and mass spectrometry for microorganism identification: Proteome-wide post-translational modifications and database search algorithms for characterization of intact H. pylori. Anal. Chem. 73, 4566–4573.
- 4) Demirev, P.A., Ho, Y.P., Ryzhov, V. and Fenselau, C. (1999) Microorganism identification by mass spectrometry and protein database searches. Anal. Chem. 71, 2732–2738.
- 5) Ryzhov, V. and Fenselau, C. (2001) Characterization of the protein subset desorbed by MALDI from whole bacterial cells. Anal. Chem. 73, 746–750.
- 6) Pineda, F.J., Antoine, M.D., Demirev, P.A., Feldman, A.B., Jackman, J., Longenecker, M. (2003) Microorganism identification by matrix-assisted laser/desorption ionization mass spectrometry and model-derived ribosomal protein biomarkers. Anal. Chem. 75, 3817–3822.
- 7) Sun, L., Teramoto, K., Sato, H., Torimura, M., Tao, H. and Shintani, T. (2006) Characterization of ribosomal proteins as biomarkers for matrix-assisted laser desorption/ionization mass spectral identification of Lactobacillus plantarum. Rapid Commun. Mass Spectrom. 20, 3789–3798.
- 8) Teramoto, K., Sato, H., Sun, L., Torimura, M. and Tao, H. (2007) A simple intact protein analysis by MALDI-MS for characterization of ribosomal proteins of two genome-sequenced lactic acid bacteria and verification of their amino acid sequences. J. Proteome Res. 6, 3899–3907.
- 9) Teramoto, K., Sato, H., Sun, L., Torimura, M., Tao, H., Yoshikawa, H. (2007) Phylogenetic classification of Pseudomonas putida strains by MALDI-MS using ribosomal subunit proteins as biomarkers. Anal. Chem. 79, 8712–8719.
- 10) Teramoto, K., Kitagawa, W., Sato, H., Torimura, M., Tamura, T. and Tao, H. (2009) Phylogenetic analysis of Rhodococcus erythropolis based on the variation of ribosomal proteins as observed by matrix-assisted laser desorption ionization-mass spectrometry without using genome information. J. Biosci. Bioeng. 108, 348–353.
- 11) Sato, H., Teramoto, K., Ishii, Y., Watanabe, K. and Benno, Y. (2011) Ribosomal protein profiling by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry for phylogenety-based subspecies resolution of Bifidobacterium longum. Syst. Appl. Microbiol. 34, 76–80.
- 12) Cogen, A.L., Nizet, V. and Gallo, R.L. (2008) Skin microbiota: A source of disease or defence? Br. J. Dermatol. 158, 442–455.
- 13) Grice, E.A., Kong, H.H., Conlan, S., Deming, C.B., Davis, J. and Young, A.C. (2009) Topographical and temporal diversity of the human skin microbiome. Science 324, 1190–1192.
- 14) Johnson, J.L. and Cummins, C.S. (1972) Cell wall composition and deoxyribonucleic acid similarities among the anaerobic coryneforms, classical propionibacteria, and strains of Arachnia propionica. J. Bacteriol. 109, 1047–1066.
- 15) McDowell, A., Perry, A.L., Lambert, P.A. and Patrick, S. (2008) A new phylogenetic group of Propionibacterium acnes. J. Med. Microbiol. 57, 218–224.
- 16) McDowell, A., Valanne, S., Ramage, G., Tunney, M.M., Glenn, J.V., McLorinan, G.C. (2005) Propionibacterium acnes types I and II represent phylogenetically distinct groups. J. Clin. Microbiol. 43, 326–334.
- 17) Dekio, I., Culak, R., Misra, R., Gaulton, T., Fang, M., Sakamoto, M. (2015) Dissecting the taxonomic heterogeneity within Propionibacterium acnes: Proposal for Propionibacterium acnes subsp. acnes subsp. nov. and Propionibacterium acnes subsp. elongatum subsp. nov. Int. J. Syst. Evol. Microbiol. 65, 4776–4787.
- 18) Oren, A. and Garrity, G.M. (2018) List of new names and new combinations previously effectively, but not validly, published. Int. J. Syst. Evol. Microbiol. 68, 3379–3393.
- 19) Nouioui, I., Carro, L., García-López, M., Meier-Kolthoff, J.P., Woyke, T., Kyrpides, N.C. (2018) Genome-based taxonomic classification of the phylum Actinobacteria. Front. Microbiol. 9, 2007.
- 20) Dekio, I., McDowell, A., Sakamoto, M., Tomida, S. and Ohkuma, M. (2019) Proposal of new combination, Cutibacterium acnes subsp. elongatum comb. nov., and emended descriptions of the genus Cutibacterium, Cutibacterium acnes subsp. acnes and Cutibacterium acnes subsp. defendens. Int. J. Syst. Evol. Microbiol. 69, 1087–1092.
- 21) Dréno, B., Pécastaings, S., Corvec, S., Veraldi, S., Khammari, A. and Roques, C. (2018) Cutibacterium acnes (Propionibacterium acnes) and acne vulgaris: A brief look at the latest updates. J. Eur. Acad. Dermatol. Venereol. 32 (Suppl 2), 5–14.
- 22) Beylot, C., Auffret, N., Poli, F., Claudel, J., Leccia, M., Del Giudice, P. (2014) Propionibacterium acnes: An update on its role in the pathogenesis of acne. J. Eur. Acad. Dermatol. Venereol. 28, 271–278.
- 23) de Brouwer, B., Veltkamp, M., Wauters, C.A., Grutters, J.C. and Janssen, R. (2015) Propionibacterium acnes isolated from lymph nodes of patients with sarcoidosis. Sarcoidosis Vasc. Diffuse Lung Dis. 32, 271–274.
- 24) Davidsson, S., Mölling, P., Rider, J.R., Unemo, M., Karlsson, M.G., Carlsson, J. (2016) Frequency and typing of Propionibacterium acnes in prostate tissue obtained from men with and without prostate cancer. Infect. Agent. Cancer 11, 26.
- 25) Aubin, G.G., Baud’huin, M., Lavigne, J.P., Brion, R., Gouin, F., Lepelletier, D. (2017) Interaction of Cutibacterium (formerly Propionibacterium) acnes with bone cells: A step toward understanding bone and joint infection development. Sci. Rep. 7, 42918.
- 26) Bémer, P., Corvec, S., Tariel, S., Asseray, N., Boutoille, D., Langlois, C. (2008) Significance of Propionibacterium acnes-positive samples in spinal instrumentation. Spine 33, E971–E976.
- 27) Tomida, S., Nguyen, L., Chiu, B.H., Liu, J., Sodergren, E., Weinstock, G.M. (2013) Pan-genome and comparative genome analyses of propionibacterium acnes reveal its genomic diversity in the healthy and diseased human skin microbiome. MBio 4, e00003–e00013.
- 28) Barnard, E., Nagy, I., Hunyadkurti, J., Patrick, S. and McDowell, A. (2015) Multiplex touchdown PCR for rapid typing of the opportunistic pathogen Propionibacterium acnes. J. Clin. Microbiol. 53, 1149–1155.
- 29) Scholz, C.F., Jensen, A., Lomholt, H.B., Bruggemann, H. and Kilian, M. (2014) A novel high-resolution single locus sequence typing scheme for mixed populations of Propionibacterium acnes in vivo. PLoS One 9, e104199.
- 30) McDowell, A., Barnard, E., Nagy, I., Gao, A., Tomida, S., Li, H. (2012) An expanded multilocus sequence typing scheme for propionibacterium acnes: Investigation of ‘pathogenic’, ‘commensal’ and antibiotic resistant strains. PLoS One 7, e41480.
- 31) Lomholt, H.B. and Kilian, M. (2010) Population genetic analysis of Propionibacterium acnes identifies a subpopulation and epidemic clones associated with acne. PLoS One 5, e12277.
- 32) Nagy, E., Urbán, E., Becker, S., Kostrzewa, M., Vörös, A., Hunyadkürti, J. (2013) MALDI-TOF MS fingerprinting facilitates rapid discrimination of phylotypes I, II and III of Propionibacterium acnes. Anaerobe 20, 20–26.
- 33) Dekio, I., Culak, R., Fang, M., Ball, G., Gharbia, S., Shah, H.N. (2013) Correlation between phylogroups and intracellular proteomes of Propionibacterium acnes and differences in the protein expression profiles between anaerobically and aerobically grown cells. BioMed Res. Int. 2013, 151797.
- 34) Amiri-Eliasi, B. and Fenselau, C. (2001) Characterization of protein biomarkers desorbed by MALDI from whole fungal cells. Anal. Chem. 73, 5228–5231.
- 35) Theel, E.S., Schmitt, B.H., Hall, L., Cunningham, S.A., Walchak, R.C., Patel, R. (2012) Formic acid-based direct, on-plate testing of yeast and Corynebacterium species by Bruker Biotyper matrix-assisted laser desorption ionization-time of flight mass spectrometry. J. Clin. Microbiol. 50, 3093–3095.
- 36) Ohta, Y., Iwamoto, S., Kawabata, S., Tanimura, R. and Tanaka, K. (2014) Salt tolerance enhancement of liquid chromatography-matrix-assisted laser desorption/ionization-mass spectrometry using matrix additive methylenediphosphonic acid. Mass Spectrom. (Tokyo) 3, A0031.
- 37) Bayston, R., Ashraf, W., Barker-Davies, R., Tucker, E., Clement, R., Clayton, J. (2007) Biofilm formation by Propionibacterium acnes on biomaterials in vitro and in vivo: Impact on diagnosis and treatment. J. Biomed. Mater. Res. A 81, 705–709.
- 38) Achermann, Y., Goldstein, E.J., Coenye, T. and Shirtliff, M.E. (2014) Propionibacterium acnes: From commensal to opportunistic biofilm-associated implant pathogen. Clin. Microbiol. Rev. 27, 419–440.
- 39) Ramage, G., Tunney, M.M., Patrick, S., Gorman, S.P. and Nixon, J.R. (2003) Formation of Propionibacterium acnes biofilms on orthopaedic biomaterials and their susceptibility to antimicrobials. Biomaterials 24, 3221–3227.
- 40) Nakamura, S., Sato, H., Tanaka, R., Kusuya, Y., Takahashi, H. and Yaguchi, T. (2017) Ribosomal subunit protein typing using matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF MS) for the identification and discrimination of Aspergillus species. BMC Microbiol. 17, 100.
- 41) Nakamura, S., Sato, H., Tanaka, R. and Yaguchi, T. (2016) Verification of ribosomal proteins of Aspergillus fumigatus for use as biomarkers in MALDI-TOF MS identification. Mass Spectrom. (Tokyo) 5, A0049.
- 42) Dekio, I., Rajendram, D., Morita, E., Gharbia, S. and Shah, H.N. (2012) Genetic diversity of Propionibacterium acnes strains isolated from human skin in Japan and comparison with their distribution in Europe. J. Med. Microbiol. 61, 622–630.
- 43) Yu, Y., Champer, J. and Kim, J. (2015) Analysis of the surface, secreted, and intracellular proteome of Propionibacterium acnes. EuPA Open Proteom. 9, 1–7.
