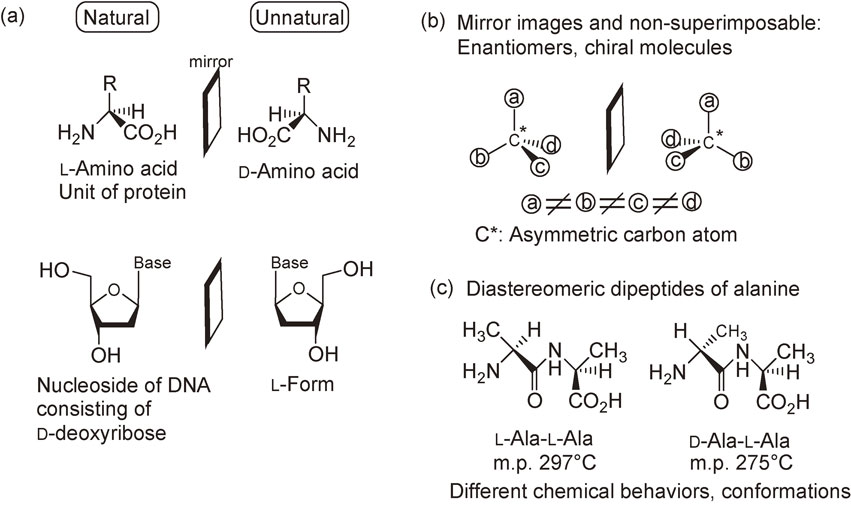
2. Asymmetric autocatalysis
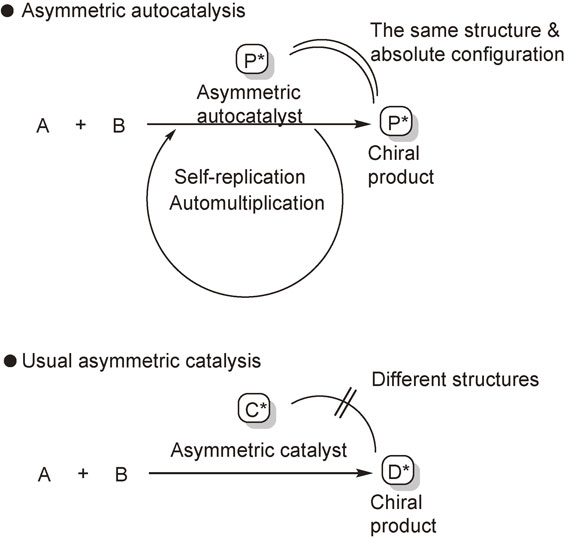
Asymmetric autocatalysis is a reaction in which a chiral product acts as a chiral catalyst for its own production (Scheme 2). The process is an automultiplication of the chiral molecule. Asymmetric autocatalysis has the following advantages over nonautocatalytic asymmetric catalysis: (1) the process involves automultiplication of a molecule; hence, the efficiency is high, (2) the new catalyst is produced as a product and the amount of catalyst therefore increases during the reaction; thus, the catalytic activity does not deteriorate, and (3) because the structures of the catalyst and product are identical, separation of the product from the catalyst is not required.
In 1953, Frank proposed a mechanism for asymmetric autocatalysis without mentioning any chemical structure.21) However, no actual experimental asymmetric autocatalysis was known until our findings of asymmetric autocatalysis of pyridyl alkanol in 1990 and pyrimidyl alkanol in 1995.22)
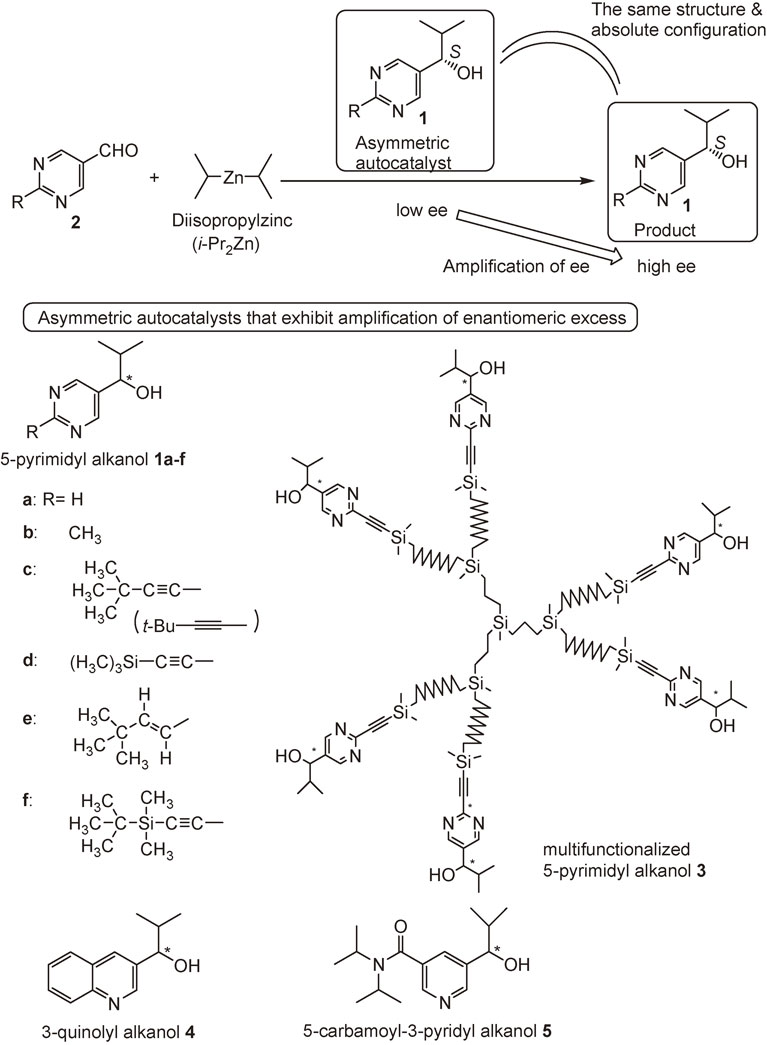
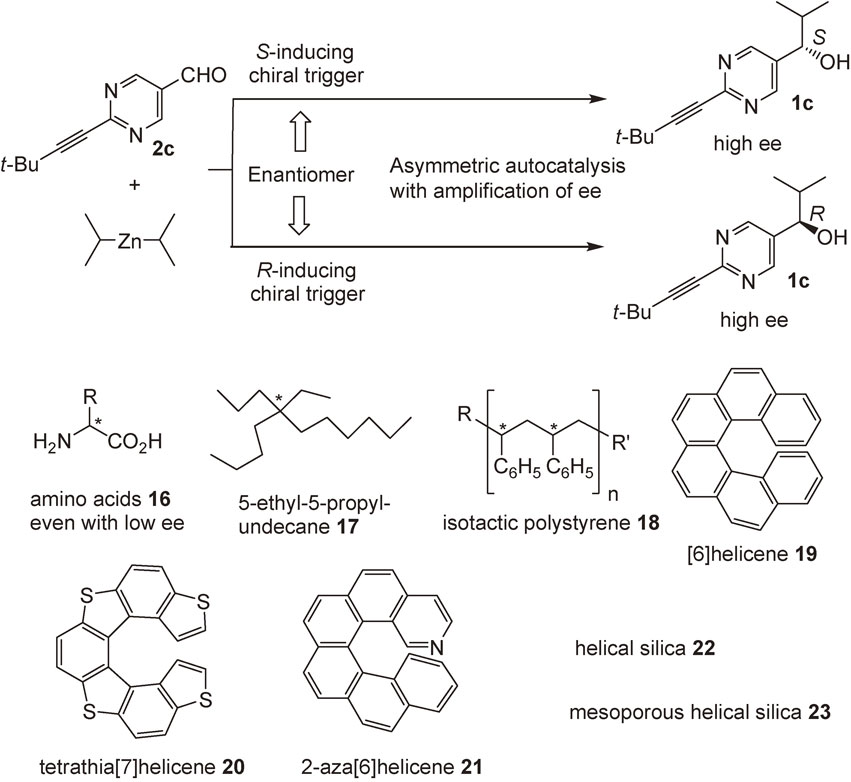
In this review, our discovery of asymmetric autocatalysis with amplification of enantiomeric excess of pyrimidyl alkanol 1,22)–26) multifunctionalized pyrimidyl alkanol 3,27),28) 3-quinolyl alkanol 4,29)–31) and 5-carbamoyl-3-pyridyl alkanol 532),33) is described (Scheme 3). The origins of homochirality of organic compounds examined by using asymmetric autocatalysis is also described.34)–41)
2.1. Background: catalytic asymmetric addition reaction of dialkylzincs to aldehydes.
Dialkylzinc reagents react with various aldehydes in the presence of chiral amino alcohols as catalysts to afford sec-alcohols with high ee.42),43)
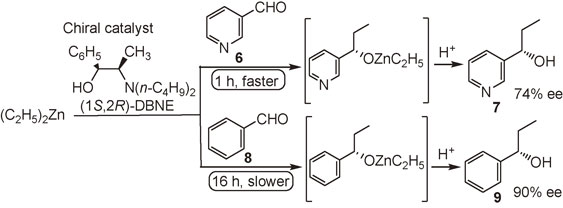
Pyridine-3-carbaldehyde 6, an aldehyde containing a nitrogen atom,44) reacts faster (1 h) than benzaldehyde 8 (16 h) with diethylzinc (Et2Zn) to afford enantioenriched 3-pyridyl alkanol 7 in a reaction in which the same chiral catalyst, i.e., (1S,2R)-N,N-dibutylnorephedrine (DBNE), is used (Scheme 4).45),46) The result showed that ethylzinc alkoxide of 3-pyridyl alkanol 7, i.e., product in situ, catalyzed the reaction.
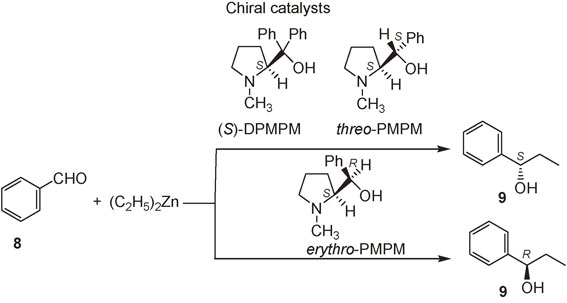
(S)-N-Methyldiphenylprolinol (DPMPM) catalyzes the addition of Et2Zn to aldehyde 8 to afford (S)-alkanol 9 with 97% ee. The catalyst (S,S)-threo-N-methylphenylprolinol (PMPM) affords (S)-alkanol 9, while (S,R)-erythro-PMPM affords (R)-alkanol 9 (Scheme 5).47) These results show that the sense of enantioselectivity is controlled by the absolute configurations of the stereogenic centers of the alcohol moieties of the catalysts threo- and erythro-PMPM.
These observations indicate that if a suitable nitrogen containing aldehyde is used, with a suitable nitrogen containing alcohol as a chiral catalyst, then the reaction with dialkylzinc might afford a chiral nitrogen containing sec-alcohol that possesses the same structure and the same absolute configuration as the catalyst. In this context, asymmetric autocatalysis is invoked.
2.2. The first asymmetric autocatalysis.
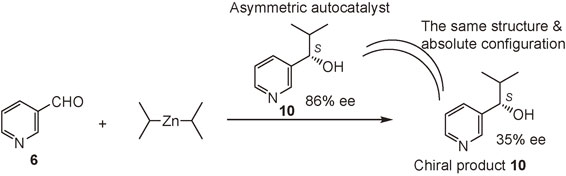
(S)-Pyridyl alkanol 10 with 86% ee acts as an asymmetric autocatalyst in the enantioselective addition of diisopropylzinc (i-Pr2Zn) to pyridine-3-carbaldehyde 6 to afford the same (S)-alkanol 10 with 35% ee (Scheme 6).48) Although the ee of the product is lower than the original catalyst, this stands as the first example of asymmetric autocatalysis, i.e., catalytic asymmetric replication of an organic compound.
2.3. Asymmetric autocatalysis with amplification of enantiomeric excess.
In 1995, we found asymmetric autocatalysis of pyrimidyl alkanol 1 with amplification of ee. (S)-Pyrimidyl alkanol 1a acts as an asymmetric autocatalyst with amplification of ee in the enantioselective addition of i-Pr2Zn to pyrimidine-5-carbaldehyde 2a to produce more of itself with the same absolute configuration (Scheme 3).22) Starting from (S)-1a with 2% ee, the product (and the initial catalyst) was used as an asymmetric autocatalyst for the next round. The ee (S)-1a reached 88% after four consecutive asymmetric autocatalyses with amplification of ee.
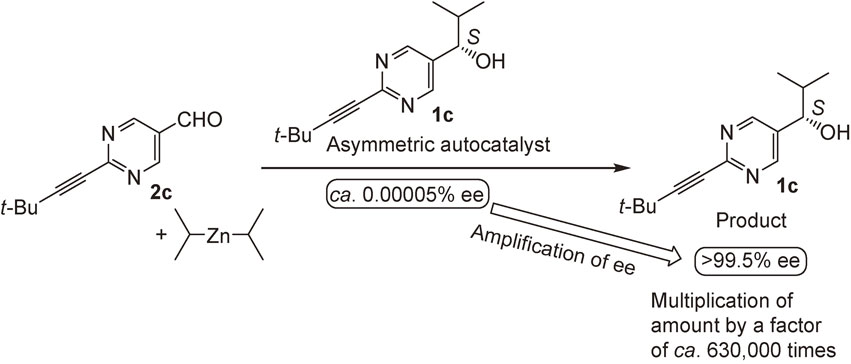
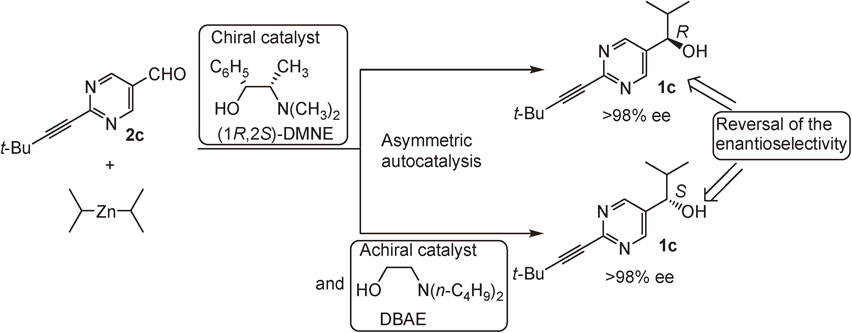
The effect of the substituent at the 2-position of pyrimidyl alkanol 1 was examined. More significant asymmetric autocatalysis was observed with pyrimidyl alkanol 1c with alkynyl substituent at the 2-position of pyrimidine ring. When (S)-pyrimidyl alkanol 1c with >99.5% ee was used as an asymmetric autocatalyst in the reaction between aldehyde 2c and i-Pr2Zn, the same alkanol 1c with >99.5% ee was obtained in >99% yield (Scheme 7).24) The mixture of the initial catalyst 1c and the product 1c was used as an asymmetric autocatalyst for the next round. Even after the 10th round, the catalytic reactivity (yield >99%) and enantioselectivity (>99.5% ee) of alkanol 1c did not decrease. Thus, this is a practically perfect asymmetric autocatalysis of pyrimidyl alkanol 1c.
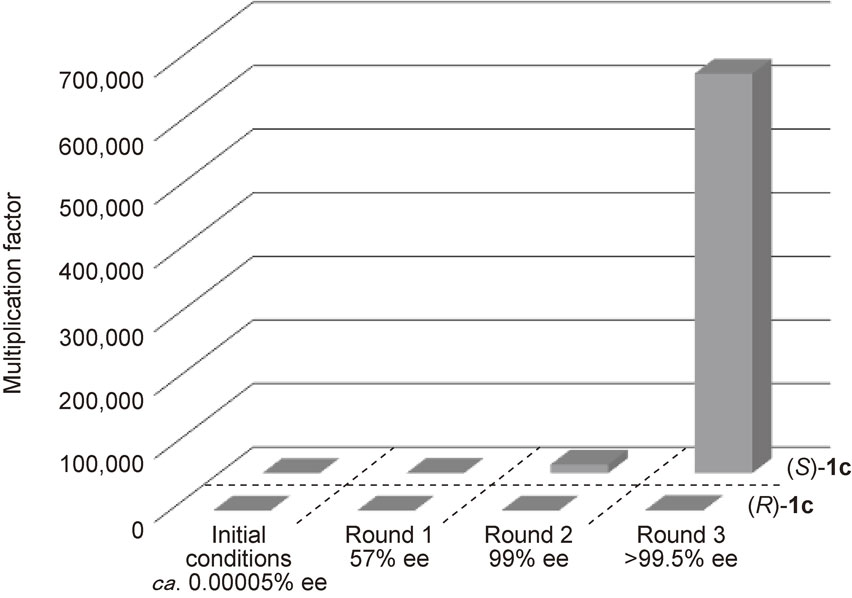
Remarkable amplification of ee was observed in the asymmetric autocatalysis of pyrimidyl alkanol 1c (Scheme 8 and Fig. 2). When it was started from an asymmetric autocatalyst 1c with an extremely low ee of ca. 0.00005%, only three consecutive asymmetric autocatalysis reactions amplified the ee of the pyrimidyl alkanol 1c to >99.5% and the amount of alkanol 1c by a factor of ca. 630,000 times.25)
It should be noted that this amplification of ee was achieved without the need for any chiral auxiliary other than the initial tiny enantiomeric imbalance of the asymmetric autocatalyst itself, thereby proving the existence of an actual chemical reaction in which very slight enantioenrichment can be amplified significantly to almost enantiopure (>99.5% ee) by the mechanism of asymmetric autocatalysis.
2.4. Mechanistic insights into asymmetric autocatalysis of 5-pyrimidyl alkanol.
The automultiplication of a chiral compound and the enormous amplification of ee are the most significant features of the asymmetric autocatalysis of 5-pyrimidyl alkanol. The enormous amplification of ee in asymmetric autocatalysis is spectacular. Thus, mechanistic insights into the asymmetric autocatalysis have attracted great attention.
We observed the relationship between the reaction time and yield of the product in the asymmetric autocatalytic reaction using an enantiopure pyrimidyl alkanol 1c.49) The result exhibited a sigmoidal curve of product formation. We also reported the kinetic analysis of asymmetric autocatalysis of pyrimidyl alkanol 1c with various ee by using chiral HPLC.50) These kinetic analyses revealed the dimeric structure of the catalytic species when the ee of the initial asymmetric autocatalyst is moderate or high based on the analogy of nonautocatalytic asymmetric dialkylzinc addition to aldehydes.51) However, when the ee of the initial asymmetric autocatalyst is low, the actual rate of amplification of ee was more significant than that simulated by the dimeric model. Thus, more aggregation of the catalyst than the dimer was envisaged.50)
Several groups examined the mechanism and modeling of asymmetric autocatalysis of pyrimidyl alkanol. Early kinetic analysis by measurement with a microcalorimeter proposed the dimeric catalyst model.52) Direct observation of the reaction solution by NMR proposed the presence of dimeric and tetrameric species.53),54) The DFT calculation study proposed the structure of catalyst aggregates.55)–57) Reaction models of spontaneous symmetry breaking, i.e. absolute asymmetric synthesis, have been proposed. These approaches also proposed the mechanisms of asymmetric autocatalysis of pyrimidyl alkanol.58)–65)
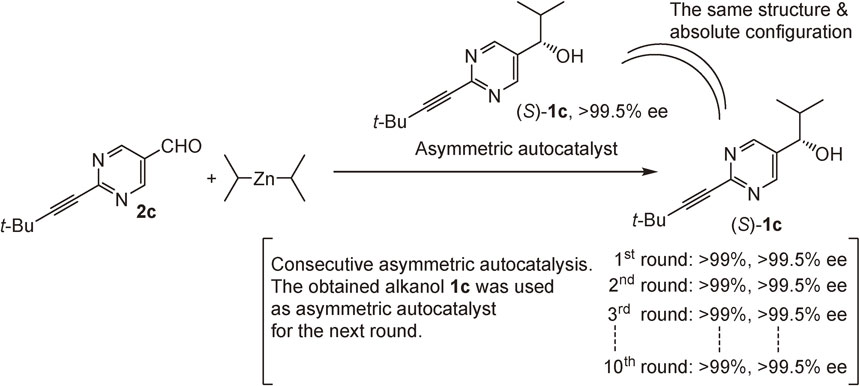
We found that the crystal structures of asymmetric autocatalysts 1c are either tetrameric or oligomeric, as determined by X-ray diffraction (Fig. 3).66),67) The tetramer structure of an asymmetric autocatalyst was determined by single-crystal X-ray diffraction analysis. Two square (Zn2O2) dimers are bridged by one 12-membered macrocycle structure with the coordination of 6 or 4 mol of i-Pr2Zn to the nitrogen atoms of pyrimidine rings (Fig. 3c). Both enantiopure (Fig. 3a) and racemic (Fig. 3b) pyrimidyl alkanols form the tetrameric crystal structure. However, these conformations differ from each other. In the enantiopure tetrameric crystal, two square (Zn2O2) dimers are positioned on the same side of the macrocycle (Fig. 3a). In the racemic tetramer, by contrast, square dimers (Zn2O2) are positioned on opposite sides (Fig. 3b). Consequently, the enantiopure tetrameric structure is more sterically crowded than the racemic tetramer. Each Zn2O2 square has a coordinatively unsaturated zinc atom (Fig. 3c). When a lower amount of i-Pr2Zn was used, higher oligomeric species crystallized.
In asymmetric autocatalysis, along with the propagation of the reaction, the concentration of the asymmetric autocatalyst increases and the concentration of i-Pr2Zn and aldehyde decreases. Thus, it is probable that the aggregation structures of the asymmetric autocatalyst are not the same between the early and final stages of the reaction. Although the crystal structures do not display the solution structures directly, structural information on the crystals provides useful insight to assist in better understanding the mechanism of asymmetric autocatalysis with amplification of ee.
Furthermore, NMR studies suggest the existence of an acetal transition state involving an acetal moiety.54) DFT calculations were used to investigate a mechanism that involved an acetal.68) Very recent reaction modeling showed that the tetramer or higher order aggregates work effectively for the amplification of chirality.69)
Although the entire reaction pathway has not yet been fully clarified, according to studies being carried out by various groups, the mechanism of asymmetric autocatalysis should be clarified in the near future.
3. Examination of the origins of chirality by using asymmetric autocatalysis with amplification of chirality
The origin of homochirality has been a long-standing problem and has attracted broad interests from many scientists.2)–10) In most cases, the ee induced by the proposed theories of the origin of chirality has been very low. Moreover, in some of the proposed theories, the induced enantioenrichments are below detection level. The linkage between the induced small enantioenrichment and the enantiopure compounds has thus remained a puzzle.11)–20)
As described in the preceding section, pyrimidyl alkanol 1 acts as an asymmetric autocatalyst with significant amplification of ee. If a proposed theory of origin of chirality could induce a small enantiomeric imbalance in pyrimidyl alkanol, either directly or indirectly, then the subsequent asymmetric autocatalysis would amplify the ee to almost enantiopure. Using asymmetric autocatalysis with amplification of ee, the theories of the origin of chirality were examined.
3.1. Circularly polarized light.
Left (l)- and right (r)-circularly polarized light (CPL) has been proposed as the origin of chirality of organic compounds such as amino acids.5) The occurrence of strong CPL is observed in star formation regions.70) It is known that the asymmetric photolysis of racemic leucine and asymmetric photosynthesis of [6]helicene by the irradiation of CPL induce enantioenrichment with only ca. 2% ee.5) However, this low ee has not been correlated to the high ee of organic compounds. We reported that leucine with low 2% ee acts as a chiral trigger of asymmetric autocatalysis to afford the pyrimidyl alkanol of higher ee with corresponding absolute configurations.71)
Direct irradiation by CPL of the racemic (rac) pyrimidyl alkanol 1c, i.e., asymmetric autocatalyst, was examined. Rac-1c was irradiated with l-CPL, and the subsequent asymmetric autocatalysis afforded highly enantioenriched (S)-alkanol 1c with >99.5% ee (Scheme 9).72) In contrast, irradiation with r-CPL in conjunction with asymmetric autocatalysis afforded (R)-1c with >99.5% ee.
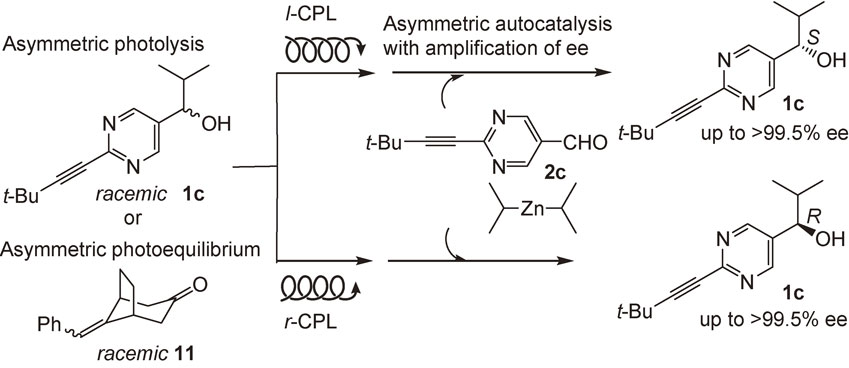
The correlation between the chirality of the CPL and (R)- or (S)-1c can be rationalized as follows. In their circular dichroism (CD) spectra at 313 nm, (R)-1c and (S)-1c exhibit positive and negative Cotton effects, respectively. Therefore, the direct irradiation of l-CPL to rac-1c would induce the asymmetric photolysis of (R)-1c in preference. Then, the remaining greater amount of (S)-1c acts as an asymmetric autocatalyst with amplification of chirality to afford (S)-1c with high ee. In this overall process, a direct correlation was achieved for the first time between the chirality of the CPL and that of an organic compound with a very high ee. Asymmetric photoequilibrium of racemic olefin 11 with the CPL in conjunction with asymmetric autocatalysis also afforded pyrimidyl alkanol 1c with high ee.73)
3.2. Chiral inorganic crystals such as quartz and cinnabar as the origin of chirality.
Certain natural minerals are known to occur as chiral crystals. Rotation of plane-polarized light, i.e., optical activity, was first found in a quartz sample. Quartz exhibits enantiomorphism and is a chiral mineral; therefore, it has long been discussed as the possible origin of chirality of organic compounds. Quartz has been used to induce enantioenrichment of organic compounds, but no significant asymmetric induction using quartz has been reported.
Asymmetric autocatalysis was examined using d- and l-quartz as chiral triggers.74) When pyrimidine-5-carbaldehyde 2c and i-Pr2Zn were reacted in the presence of d-quartz powder, (S)-pyrimidyl alkanol 1c with 97% ee was obtained in 95% yield (Scheme 10). By contrast, in the presence of l-quartz, asymmetric autocatalysis was triggered to afford (R)-alkanol 1c with 97% ee in 97% yield. These results clearly indicate that quartz acts as the origin of chirality, in conjunction with asymmetric autocatalysis, to afford alkanols 1c with high ee and with absolute configurations correlated with those of quartz. A low ee induced in the initially formed zinc alkoxide of pyrimidyl alkanol (induced by chiral quartz) was amplified significantly by subsequent asymmetric autocatalyses to afford 1c with very high ee. Thus, the chirality of an inorganic crystal was correlated, for the first time, to the chirality of an organic compound with very high ee.
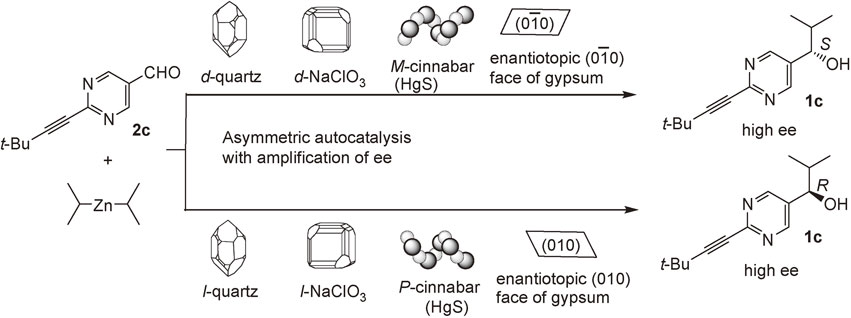
Sodium chlorate (NaClO3) and sodium bromate (NaBrO3) are chiral inorganic ionic crystals. Asymmetric autocatalysis using d-NaClO3 as a chiral trigger afforded (S)-alkanol 1c with high ee, while l-NaClO3 afforded (R)-1c.75) On the other hand, asymmetric autocatalysis in the presence of d-NaBrO3 afforded (R)-1c, while l-NaBrO3 afforded (S)-1c.76) These correlations are in agreement with the fact that the signs of optical activity for NaClO3 and NaBrO3 in the same enantiomorph are opposite. Thus, in this asymmetric autocatalysis, the crystals in the same enantiomorph have the same sense of enantioselectivity.
Cinnabar is a red-colored chiral helical crystal of mercury(II) sulfide (α-HgS) that has been used as a pigment for seal stamps and paint, and as a traditional Chinese medicine. Enantiomorphous P- and M-crystals of cinnabar are constructed with –S–Hg–S–Hg– spiral chains. It was found that P-HgS triggered asymmetric autocatalysis to afford (R)-1c with high ee.77) On the other hand, M-HgS afforded (S)-1c with high ee. Retgersite (nickel sulfate hexahydrate) is a naturally occurring mineral that exhibits enantiomorphism. It was also found that retgersite acts as a chiral trigger of asymmetric autocatalysis.78)
Gypsum (calcium sulfate dihydrate) is a common mineral. It has been used for sculptures, plaster boards in buildings, and plaster casts for medical use. Although the crystal structure of gypsum is achiral, gypsum exhibits the two-dimensional enantiotopic cleavage (010) and (0–10) face that readily appears upon slicing of the crystal. Pyrimidine-5-carbaldehyde 2c was placed on the enantiotopic (010) face of gypsum and exposed to the vapor of i-Pr2Zn.79) (R)-Alkanol 1c was formed. The ee of 1c was amplified by the subsequent asymmetric autocatalyses. The reaction on the opposite enantiotopic (0–10) face afforded (S)-alkanol 1c. Thus, the enantiotopic surface of an achiral inorganic crystal was found to act as a chiral trigger of asymmetric autocatalysis.
As described, in conjunction with asymmetric autocatalysis, chiral inorganic crystals and the enantiotopic face of an achiral inorganic crystal act as the origins of chirality to afford organic compounds with the correlated absolute configurations with high ee.
3.3. Chiral crystals composed of achiral organic compounds.
Certain classes of achiral organic compounds are known to form chiral crystals. The use of these chiral organic crystals as reactants in certain stereospecific reactions has been reported. However, chiral organic crystals consisting of achiral organic compounds have rarely been used as chiral catalysts or initiators in an enantioselective reaction. Investigations were therefore carried out to determine whether chiral crystals composed of achiral organic compounds could act as chiral triggers in asymmetric autocatalysis (Scheme 11).
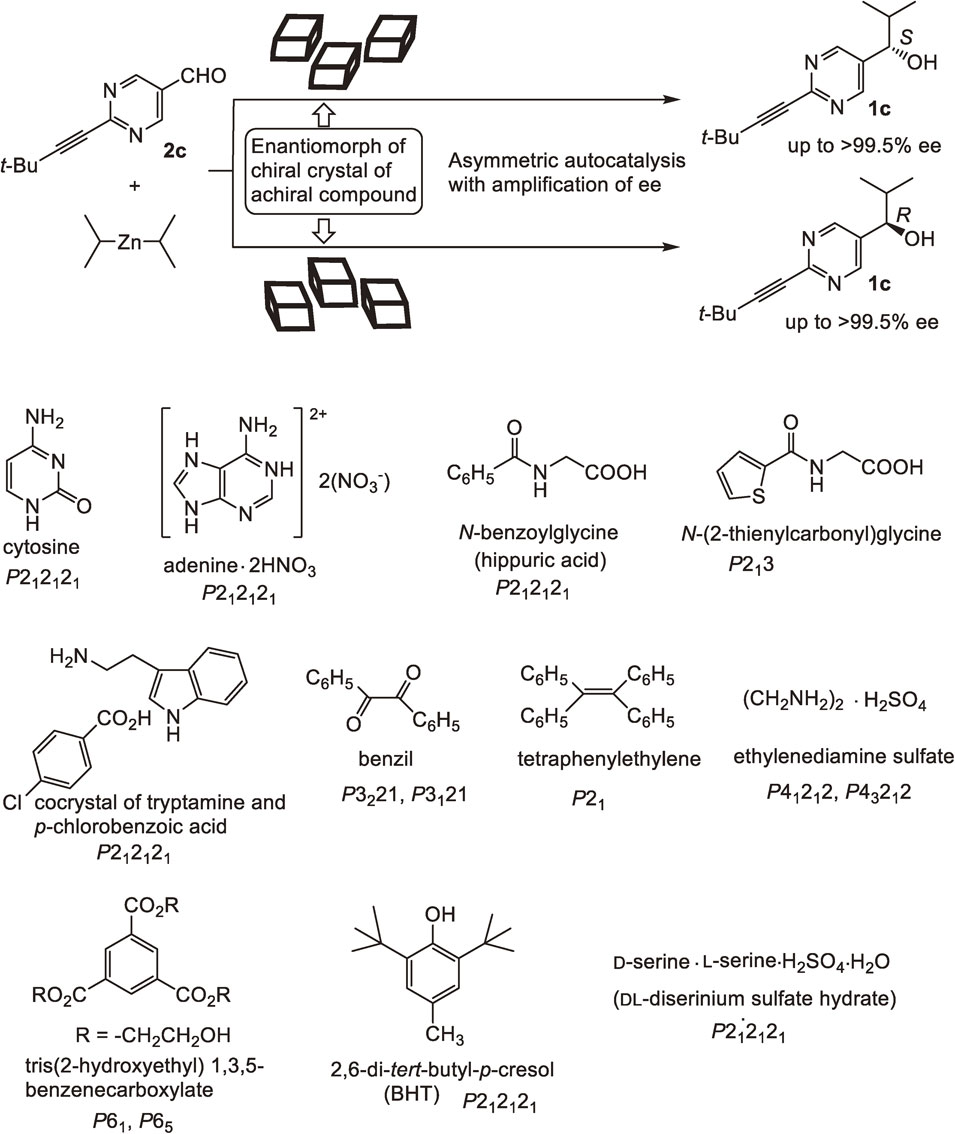
Cytosine is an achiral compound and an achiral nucleobase that is formed under prebiotic conditions. Crystallization from methanol affords chiral crystals that exhibit either plus or minus Cotton effects in solid-state CD spectra at ca. 310 nm. Chiral crystals of cytosine have been used as chiral triggers for asymmetric autocatalysis. Using the [CD(+)310Nujol]-cytosine crystal as a chiral trigger, the reaction between aldehyde 2c and i-Pr2Zn afforded enantioenriched (R)-alkanol 1c with 99.5% ee in conjunction with asymmetric autocatalysis with amplification of ee.80) On the other hand, in the presence of the [CD(−)310Nujol]-cytosine crystal as a chiral trigger, the reaction afforded enantioenriched (S)-1c with 99.5% ee. These results indicate that the chiral crystal of achiral cytosine acts as the chiral source of the resulting 1c.
With regard to the formation of cytosine chiral crystal, we found an unprecedented type of the origin of chirality. The achiral crystal of cytosine monohydride is known to be formed after the crystallization of cytosine from water. It was found that the elimination of the crystal water by heating one of the enantiotopic faces of an achiral crystal of cytosine monohydrate affords a chiral crystal of dehydrated cytosine.81) The chirality of the formed dehydrated cytosine crystal is correlated with the enantiotopic face of the achiral cytosine monohydrate crystal. It was also found that dehydration of crystal water under reduced pressure, instead of heating, from the same enantiotopic face affords a chiral dehydrate cytosine crystal with the opposite chirality.82) These results offer the first example of the formation of chiral crystals with controlled chirality by the dehydration of crystal water from an achiral crystal.
Adenine is an achiral nucleobase. Adenine dinitrate forms chiral crystals. Chiral crystals composed of adenine dinitrate act as chiral triggers for asymmetric autocatalysis of pyrimidyl alkanol 1c83) which indicates that achiral nucleobases such as cytosine and adenine in their chiral crystalline state act as the origins of chirality.
It was also found that enantiomorphous crystals composed of achiral N-benzoylglycine,84) N-(2-thienylcarbonyl)glycine,85) cocrystal of tryptamine and p-chlorobenzoic acid,86) benzil,87) tetraphenylethylene,88) ethylenediamine sulfate,89) benzene triester90) and 2,6-di-t-butyl-p-cresol (BHT)91) act as chiral triggers for asymmetric autocatalysis of 1c. Thus, asymmetric autocatalysis could serve as a macroscopic observation tool of molecular-level information on the structure of crystal surfaces.85)
A chiral crystal composed of a racemic organic compound can also act as a chiral trigger in asymmetric autocatalysis. The racemate of serine formed the chiral crystal (P212121) of dl-diserinium sulfate hydrate. M-Crystals of dl-diserinium sulfate hydrate triggered asymmetric autocatalysis to afford (R)-pyrimidyl alkanol 1c.92) By contrast, asymmetric autocatalysis triggered by P-crystals afforded (S)-alkanol 1c. As described, it was found that chiral crystals formed from a racemic compound acted as a chiral source.
3.4. Asymmetric autocatalysis initiated on the enantiotopic faces of achiral organic crystals of achiral organic compound.
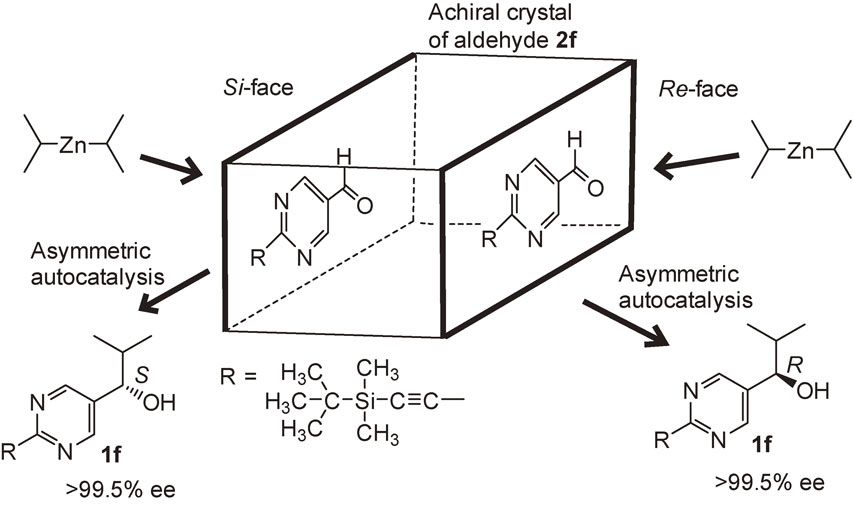
There are certain classes of achiral organic compounds that form achiral crystals with enantiotopic faces. 2-(tert-Butyldimethylsilylethynyl)pyrimidine-5-carbaldehyde 2f formed an achiral crystal (P–1) with enantiotopic faces (Scheme 12). When one of these faces was exposed to the vapor of i-Pr2Zn, the exposed Re-face of aldehyde was attacked by i-Pr2Zn to afford (R)-pyrimidyl alkanol 1f.93) By contrast, exposure of the opposite enantiotopic face (the Si-face of aldehyde) afforded (S)-alkanol 1f. The ee of the obtained alkanol was amplified to >99.5% by the subsequent asymmetric autocatalyses.
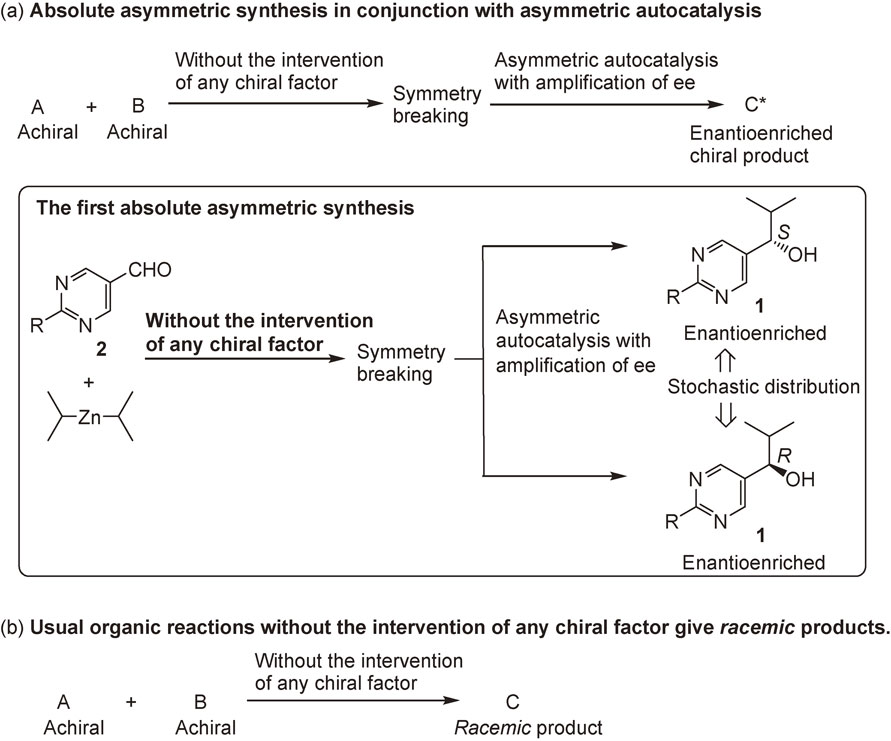
3.5. Absolute asymmetric synthesis in conjunction with asymmetric autocatalysis.
In his commentary on absolute asymmetric synthesis, Mislow termed absolute asymmetric synthesis as the formation of an enantioenriched compound from achiral compounds without the intervention of any chiral factor.3) Absolute asymmetric synthesis has been proposed as one of the origins of homochirality in nature. However, it is widely accepted that without the intervention of any chiral factor, a racemic chiral product is always formed from achiral compounds (Scheme 13b). This phenomenon is explained as follows: the probability of the formation of R and S products is equal; furthermore, the molecules are produced in huge numbers, and the ratio of R and S enantiomeric products in a reaction is 50:50 (a racemate).
However, according to the theory of statistics, the numbers of the R and S product enantiomers are rarely exactly the same, i.e., there are almost always statistical fluctuations in the numbers of produced enantiomers.94),95) For example, when a coin is flipped 100 times, the probability of exactly 50 heads and 50 tails is only 8%. In the other 92% of cases, either heads or tails will be in excess, such as 48 to 52, 51 to 49, and so on.
As described in the preceding section, asymmetric autocatalysis of pyrimidyl alkanol in the reaction between pyrimidine-5-carbaldehyde and i-Pr2Zn amplifies very low ee to >99.5% ee.25) What would happen if pyrimidine-5-carbaldehyde was reacted with i-Pr2Zn without using any chiral factor? The initial small enantioenrichment of the product, i.e., isopropylzinc alkoxide of pyrimidyl alkanol, as a result of the fluctuation in racemic mixtures, could be amplified by the subsequent asymmetric autocatalyses with amplification of ee, and could produce an enantiomerically enriched product (Scheme 13a). Thus, absolute asymmetric synthesis without the intervention of any chiral factor was examined in the reaction between pyrimidine-5-carbaldehyde and i-Pr2Zn.96),97)
The reactions of pyrimidine-5-carbaldehyde 2c and i-Pr2Zn without the intervention of any chiral factor was found to afford enantioenriched (S)- or (R)-alkanol 1c with stochastic distributions of formation (Scheme 13a). When aldehyde 2c was reacted with i-Pr2Zn, the subsequent asymmetric autocatalysis with amplification of ee afforded pyrimidyl alkanol 1c with an ee well above the detection level. The absolute configurations of 1c produced under these conditions exhibited a stochastic distribution of S and R enantiomers: (S)-1c formed 19 times and (R)-1c formed 18 times (Fig. 4).96) In addition, the reactions between pyrimidine-5-carbaldehyde and i-Pr2Zn in the presence of achiral amorphous silica gel98) and achiral amines99) without using any chiral factor afforded (S)-1c or (R)-1c in a stochastic manner. Absolute asymmetric synthesis with stochastic distribution of the product was also reported by another group using pyrimidine-5-carbaldehyde 2b and i-Pr2Zn.100)
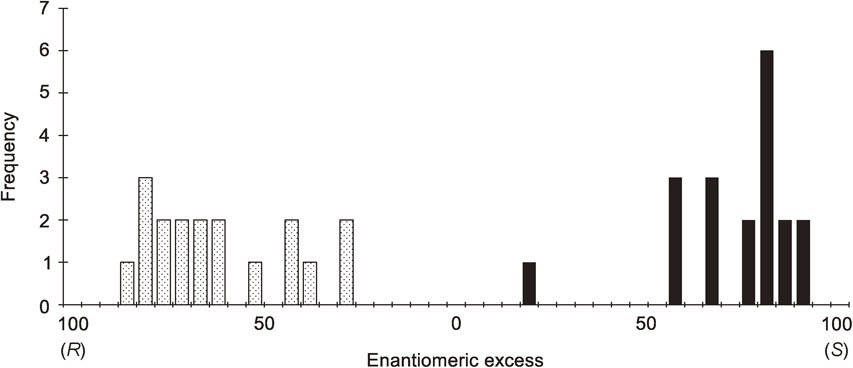
These results of stochastic behavior in the formation of enantioenriched pyrimidyl alkanol constitute one of the conditions necessary for spontaneous absolute asymmetric synthesis. It should be noted that the present absolute asymmetric synthesis is counterintuitive, considering the common knowledge that the reaction between achiral reactants without the intervention of any chiral factor always affords racemic chiral products. Thus, the present results stand as the first example of spontaneous absolute asymmetric synthesis as defined by Mislow.3)
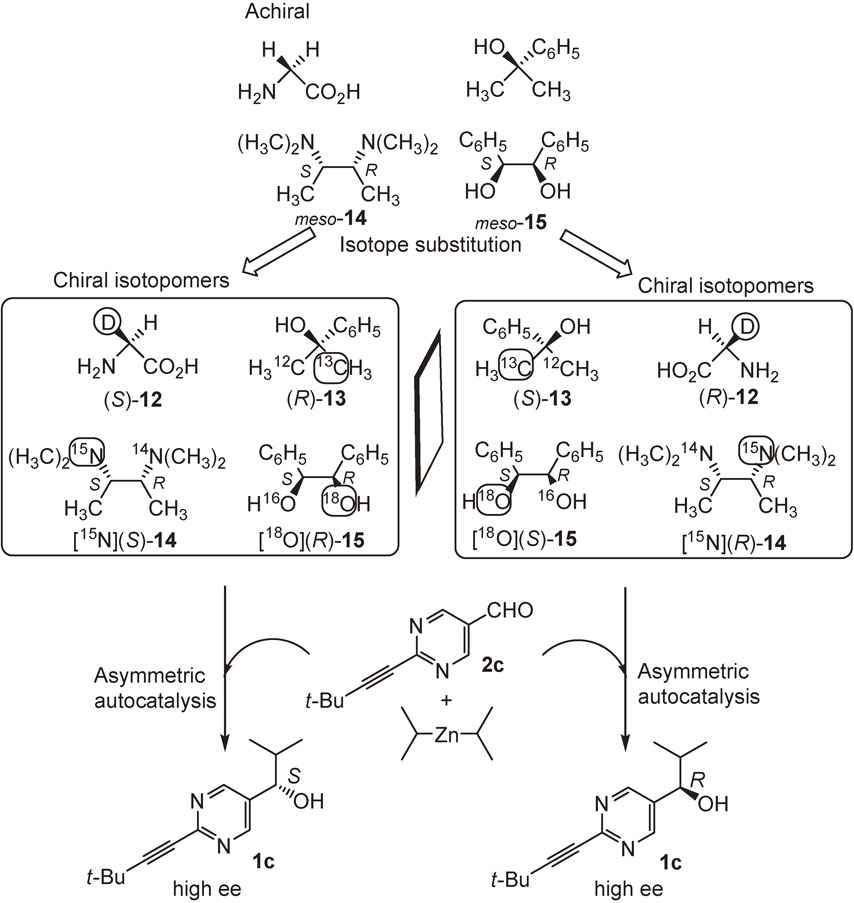
3.6. Asymmetric autocatalysis triggered by chiral compounds arising from hydrogen (D/H), carbon (13C/12C), nitrogen (15N/14N), and oxygen (18O/16O) isotopomers.
Asymmetric autocatalysis was applied to the discrimination of chirality of isotopomers. When pyrimidine-5-carbaldehyde 2c is treated with i-Pr2Zn using chiral isotopomers as chiral triggers, the sense of absolute configuration of the product 1c should be controlled by the chirality of the external chiral trigger (Scheme 14).
Glycine does not possess any asymmetric carbon atom. However, the compound becomes chiral when one of the hydrogen atoms of the methylene group of glycine is substituted by deuterium. Chiral isotopomers of glycine-α-d 12 were used as chiral triggers in an asymmetric autocatalysis in the reaction between i-Pr2Zn and pyrimidine-5-carbaldehyde 2c to afford alkanol 1c with high ee.101) The absolute configuration of the produced alkanol 1c was controlled by the chirality of the trigger resulting from hydrogen isotope substitution. When i-Pr2Zn was reacted with aldehyde 2c using a chiral trigger of (S)-glycine-α-d 12, (S)-pyrimidyl alkanol 1c with high ee was formed. By contrast, in the presence of (R)-glycine-α-d 12, the opposite (R)-1c was formed. Chiral deuterated primary alcohols102) can also trigger asymmetric autocatalysis. These results are the first examples of highly enantioselective synthesis induced by chiral compounds owing to deuterium substitution.103)
Carbon (13C/12C), nitrogen (15N/14N) and oxygen (18O/16O) isotopomers should be much more difficult to discriminate experimentally than H/D isotopomers because the chirality originates from the much smaller difference in atomic weight between carbon-13/carbon-12, nitrogen-15/nitrogen-14, and oxygen-18/oxygen-16 (Scheme 14). The induction of detectable enantioenrichment in asymmetric reactions by carbon, nitrogen, and oxygen isotopomers has not been reported earlier. Asymmetric autocatalysis was therefore examined using these carbon, nitrogen, and oxygen isotopomers as chiral triggers.
Many apparently achiral organic molecules on Earth may be chiral because of random substitution of the 1.11% naturally abundant 13C for 12C in an enantiotopic moiety within a structure. Dimethylphenylmethanol 13 is an achiral compound because it has two methyl groups on the same carbon atom. However, one of the carbon atoms of the two methyl groups is labeled with 13C; hence, the compound becomes a chiral carbon isotopomer. Asymmetric autocatalysis was conducted using the chiral carbon isotopomer of alkanol 13 as a chiral trigger.104) When i-Pr2Zn and aldehyde 2c were reacted in the presence of (R)-dimethylphenylmethanol 13, arising from 13C substitution of one of the methyl groups, (R)-pyrimidyl alkanol 1c with high ee was formed (Scheme 14). By contrast, (S)-13 triggered the formation of (S)-1c.
meso-N2,N2,N3,N3-Tetramethyl-2,3-butanediamine 14 is an achiral compound because, despite the presence of asymmetric carbon atoms, compound 14 is superimposable with its mirror image. However, when one of the nitrogen (14N) atoms is substituted by 15N, the diamine 14 becomes a chiral nitrogen (15N/14N) isotopomer 14. When asymmetric autocatalysis was triggered by [15N](R)-diamine 14, (R)-pyrimidyl alkanol 1c was formed with >99.5% ee. By contrast, the chiral trigger [15N](S)-diamine 14 afforded (S)-alkanol 1c with >99.5% ee.105) It was also found that oxygen (18O/16O) isotopomers of 1,2-diphenyl-1,2-ethanediol 15 and glycerin acted as chiral triggers of asymmetric autocatalysis.106),107)
These asymmetric inductions by carbon, nitrogen, and oxygen isotopically chiral compounds are the first examples in the research field of chirality. The neglected carbon, nitrogen, and oxygen isotope chirality of many organic compounds on Earth can therefore be discriminated by asymmetric autocatalysis with amplification of the extremely small chiral influence between 13C/12C, 15N/14N and 18O/16O.108),109)