Reviews
Molecular mechanisms of coupling to voltage sensors in voltage-evoked cellular signals
2019 Volume 95 Issue 3 Pages 111-135
Details
2019 Volume 95 Issue 3 Pages 111-135
The voltage sensor domain (VSD) has long been studied as a unique domain intrinsic to voltage-gated ion channels (VGICs). Within VGICs, the VSD is tightly coupled to the pore-gate domain (PGD) in diverse ways suitable for its specific function in each physiological context, including action potential generation, muscle contraction and relaxation, hormone and neurotransmitter secretion, and cardiac pacemaking. However, some VSD-containing proteins lack a PGD. Voltage-sensing phosphatase contains a cytoplasmic phosphoinositide phosphatase with similarity to phosphatase and tensin homolog (PTEN). Hv1, a voltage-gated proton channel, also lacks a PGD. Within Hv1, the VSD operates as a voltage sensor, gate, and pore for both proton sensing and permeation. Hv1 has a C-terminal coiled coil that mediates dimerization for cooperative gating. Recent progress in the structural biology of VGICs and VSD proteins provides insights into the principles of VSD coupling conserved among these proteins as well as the hierarchy of protein organization for voltage-evoked cell signaling.
Communicated by Masanori OTSUKA, M.J.A.
Abbreviations: Anap: 3-(6-acetylnaphthalen-2-ylamino)-2-aminopropanoic acid; CatSper: cation channel of sperm; Cav: voltage-gated Ca2+; CNG: cyclic nucleotide-gated; cryo-EM: cryoelectron microscopy; EPR: electron paramagnetic resonance; FRET: fluorescence resonance energy transfer; GEVI: gene-encoded voltage indicator; GFP: green fluorescent protein; H2O2: hydrogen peroxide; HCN channel: hyperpolarization-activated, cyclic nucleotide-gated channel; HOCl: hypochlorous acid; HVCN1: hydrogen voltage-gated channel 1 (gene name for Hv1, voltage-gated proton channel); HVRP1: Hv1-related protein 1; Kcv: virus-derived K+ channel; Kir: inward rectifier K+; Kv: voltage-gated K+; MPO: myeloperoxidase; NADPH: nicotinamide adenine dinucleotide phosphate; Nav: voltage-gated Na+; NHE: sodium proton exchanger; PBM: phosphoinositide-binding motif; PD: phosphatase domain; PGD: pore-gate domain; PHD: pleckstrin homology domain; PI: phosphoinositide; PS: pregnenolone sulfate; PtdIns(3,4)P2: phosphatidylinositol-3,4-bisphosphate; PtdIns(3,4,5)P3: phosphatidylinositol-3,4,5-trisphosphate; PtdIns(3,5)P2: phosphatidylinositol-3,5-bisphosphate; PtdIns(4,5)P2: phosphatidylinositol-4,5-bisphosphate; PtdIns4P: phosphatidylinositol-4-monophosphate; PTEN: phosphatase and tensin homolog (deleted on chromosome ten); ROS: reactive oxygen species; RyR: ryanodine receptor; SNARE complex: soluble NSF attachment protein receptor complex; TAPP1: tandem PH domain containing protein 1; TPC: two-pore channel; TRIC: trimeric intracellular channel; TRP: transient receptor potential; VCF: voltage clamp fluorometry; VGIC: voltage-gated ion channel; VSD: voltage sensor domain; VSLD: voltage-sensor like domain; VSP: voltage-sensing phosphatase.
Membrane excitation plays fundamental roles in the functions of neurons, muscle, endocrine cells, and electric organs. Na+ and K+ conductances through the membrane, mediated by voltage-gated Na+ and K+ ion (Nav and Kv) channels, respectively, were the first elements shown to underlie the membrane excitability of nerves.1) Later, voltage-gated Ca2+ (Cav) channels were identified in muscle, neurons, and endocrine cells.2),3) The molecular correlates of these ion channels were identified through studies involving biochemistry, molecular genetics,4) and molecular cloning.5),6) Proteins comprising Nav, Kv, and Cav channels share a common architecture that includes a voltage sensor domain (VSD) consisting of four transmembrane helices and a pore-gate domain (PGD) consisting of two transmembrane helices with an intervening turret region (Fig. 1). These voltage-gated ion channels (VGICs), together with related proteins, such as hyperpolarization-activated, cyclic nucleotide-gated channels (HCN channels) and two-pore channels (TPCs), are all classified as members of the VGIC superfamily.7) In Kv and HCN channels, one subunit contains a single VSD and PGD, and four subunits assemble into one channel. In Nav and Cav, one channel is formed by a long polypeptide consisting of four homologous repeats, each of which contains a single VSD and PGD. In TPCs, one channel is formed by two homologous subunits, each containing two repeats. Transient receptor potential (TRP) channels are ion channels transducing multimodal chemical and physical stimuli, such as stretch, temperature change, and oxygen concentration, into intracellular signals. The transmembrane regions of TRP channels have a molecular architecture that resembles that of VGICs, consisting of a voltage-sensor like domain (VSLD) and a PGD.
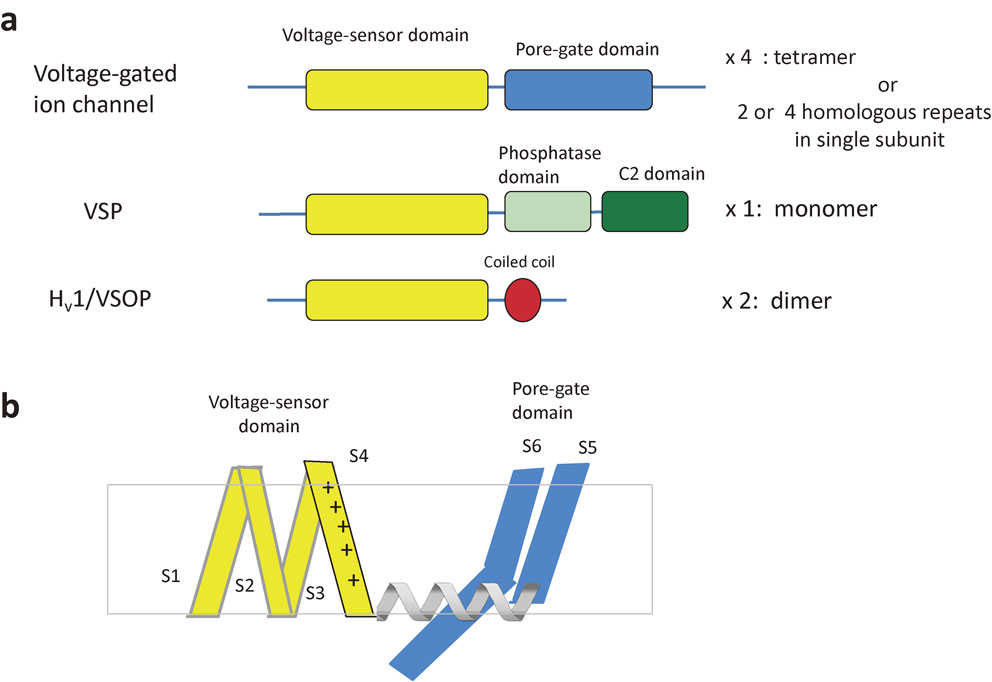
Scheme of the membrane topologies of VGIC and two VSD-containing proteins. a. VSP and Hv1. VGIC contains the voltage-sensor domain (VSD) and the pore-gate domain (PGD). Most of VGICs contain N-terminal and C-terminal cytoplasmic domains which are important for subunit assembly and gating (not specifically illustrated here). VSP has the transmembrane VSD and the cytoplasmic enzyme region which consists of the phosphatase domain and C2 domain. There is a short linker between the VSD and PD (called VSD-PD linker) (not illustrated here). Transmembrane region of Hv1, the voltage-gated proton channel, corresponds to the VSD of VGIC and VSP. Hv1 contains the cytoplasmic coiled coil region which is critical for dimer formation and gating. b. Illustration of one unit of VSD (consisting of four transmembrane helices) and PGD of domain-swapped type VGIC. Helical linker connects the two domains. See Fig. 8 for domain-nonswapped type VGIC.
The protein structures of most VGIC species have been solved using X-ray crystallography and, more recently, with single particle cryo-electron microscopy (cryo-EM) (Fig. 2).8) One remarkable finding from recent studies of the structures of VGICs is that the coupling of the VSD to the PGD is diverse and optimized for the physiological functions of each individual class of VGIC. Moreover, in several atypical VSD-containing proteins, the coupling of the VSD is not for driving a PGD. In voltage-sensing phosphatases, for example, the VSD is coupled to the enzyme and regulates phosphatase activity. In the voltage-gated proton channel, Hv1, which lacks a PGD, the VSD operates as an ion permeation pathway as well as a voltage sensor, and two VSDs interact with each other for cooperative gating. How protons selectively permeate through the VSD in Hv1 remains a mystery. In addition, cyclic nucleotide-gated (CNG) channels and many TRP channels have immobile VSDs.

Gallery of protein structures of VGICs and VSP (taken from ref. 157). Kv1.2–Kv2.1 chimera: human voltage-gated potassium channel (PDB ID: 2R9R), TPC1: plant two-pore cation channel (PDB ID: 5E1J), Cav1.1: human skeletal muscle-type voltage-gated calcium channel (PDB ID: 5GJV), NavPaS: insect voltage-gated sodium channel (PDB ID: 5X0M), HCN1: human hyperpolarization-activated cyclic nucleotide-gated channel (PDB ID: 5U6O), Slo1: large conductance calcium-activated voltage-gated potassium channel from Aplysia (PDB ID: 5TJ6), Ci-VSP: sea squirt voltage-sensing phosphatase (full length model based on coordinates from PDB ID: 3AWF and PDB ID: 4G7V), mHv1cc: mouse voltage-gated proton channel in a chimeric form containing part of S2–S3 from Ci-VSP and the coiled coil structure of GCN4 (dimer model based on coordinates from PDB ID: 3WKV). NavPaS, Kv1.2–Kv2.1 chimera and Slo1 have four VSDs within a tetramer. TPC1 has four VSDs within a dimer. Ci-VSP has a single VSD. mHv1cc has two VSDs within a dimer.
In this article, we first summarize the structure–function relationship and molecular mechanisms of coupling between the VSD and PGD in VGICs. Then, we describe VSD-effector coupling and VSD-VSD coupling in two VSD-containing proteins, voltage-sensing phosphatase (VSP) and Hv1, respectively, both of which do not contain a canonical PGD. We also state more emerging functions of the VSD-like region in other proteins and the hierarchy of voltage signals. Finally, future directions of research are discussed.
The VSD is a protein structure critical for sensing membrane voltage and is highly conserved among VGICs. It consists of four transmembrane helices, S1–S4, with S4 showing a signature pattern of amino acid alignment: positively charged residues aligned with two intervening hydrophobic residues (Fig. 3a).9) The positive charges in S4 are counterbalanced by multiple negative charges on acidic residues in the other three helices (S1–S3) to form salt bridges that are periodically aligned, like a ladder (Fig. 3b). This ladder-like alignment of salt bridges spans a constricted highly hydrophobic region. A hydrophobic residue, phenylalanine, in S2 is highly conserved among VGICs, except in some rare cases.10) This residue functions to separate two aqueous crevices, thereby providing a charge transfer center for the positive charges on S4 to translocate. Sensing an electric field across the membrane,11),12) S4 undergoes translation or helical motion against the other three helices. The charge displacement achieved through this voltage-induced conformational change in the VSD can be measured with an electrophysiological method as a macroscopic transient current (gating currents).13),14)

Amino acid sequence of S4 (a) and the X-ray crystal structure of the VSD of Ci-VSP with its membrane topology (b: taken from ref. 157).
The PGD consists of two transmembrane helices (S5 and S6) and an intervening loop and contains two major structures that regulate ion permeation. The loop between S5 and S6 forms a selectivity filter near the extracellular exit of the pore. This determines which ions can permeate through the pore. Bundled crossing of four sets of S5 and S6 near the intracellular exit of the pore forms the gate structure. A part of S6 on the side of the membrane closer to the cytoplasm contains a kinked region, which is critical for regulating gating.15)
How the VSD senses changes in transmembrane potential and what motion is induced have long been hot issues. The first X-ray crystal structure of a VGIC was that of the Kv channel from the archeabacterium Aeropyrum pernix, called KvAP.16) The crystal structure shows the S3–S4 region protruding to the external side of the protein in the solvent environment. This structure led to the hypothesis that parts of S3 and S4 and the intervening extracellular loop (S3–S4 loop) forms a paddle-like structure and undergoes substantial movement from the hydrophobic to the aqueous environment (“paddle” model).17) Later, however, the solved structure of the human Kv channel showed the position of S4 within the VSD and suggested that S4 in the crystal structure of KvAP was dislocated to the outside as a result of the crystal packing. Nevertheless, the paddle model stimulated several biophysical studies of S4 motion.18),19) Various models of S4 motion, including the transporter model, in which there is only minor helical motion of S4, and simple translation of S4 through other parts of the VSD, were proposed.20) These models differ with respect to the distance and direction of S4 motion. Later, the solved atomic structures of VGICs and biophysical measurements led to the consensus idea that the movement of S4 is characterized by a mixture of translational and helical screw motions.20) A study of long time scale (∼ ms), all-atom molecular dynamics simulations of the Kv channel upon a change in membrane potential also support such motion of S4.21)
As is evident from their names, VGICs are classified based on their biophysical nature, including ion selectivity, which is rigidly dictated by the structure of the PGD and the direction of the membrane potential that activates channel opening. Most VGICs are activated by depolarization, although HCN channels are activated by hyperpolarization. In many types of Kv, Nav, and Cav channels,22)–24) as well as TPCs, which are endosomal/lysosomal voltage-gated cation channels, the VSD is “domain-swapped” with the PGD. In other words, the VSD is linked to the PGD within the same subunit (in Kv) or repeat (in Nav and Cav), but is located next to the PGD in a neighboring subunit (or repeat) (Fig. 4, right panel). The linker between the VSD and PGD (called the S4–S5 linker) of one subunit runs beneath the pore-forming helix of another subunit before reaching the PGD of the same subunit. The S4–S5 linker is a helix forming a cuff around the S4 helix that also makes contact with the C-terminal part of S6. This category of channels with a VSD characterized as being domain-swapped includes most classically studied VGICs, including Nav, Kv, and Cav channels, which are primarily activated by membrane depolarization. Conformational changes in the VSD can be transmitted to the gate through the linker and C-terminal part of S6. It should be noted that in these types of VGICs the cytoplasmic region is not domain-swapped. For example, the T1 domain of Kv1, which is known to be involved in tetrameric assembly but not in regulating channel gating, is not domain-swapped. A subunit-assembly domain containing a coiled-coil motif in the C-terminal cytoplasmic region of Kv7 is also not domain-swapped.25)

Two categories of VGICs dependent on VSD-PGD organization: domain-swapped vs. domain-nonswapped. A figure from ref. 37 is used.
The critical functions of the S4–S5 linker and C-terminal part of S6 in VSD-PGD coupling were established in a number of studies, two of which are seminal. In one of these studies, voltage dependence was conferred to the pH-gated potassium channel from the soil bacteria Streptomyces lividans, which is called the KcsA channel (a channel that lacks a VSD) by constructing a chimeric protein with Kv1 (which contains a VSD). In that case, the transfer of not only the VSD but also the S4–S5 linker and C-terminal part of S6 helix from Kv1 was necessary to confer voltage sensitivity on the KcsA channel.26) The second seminal study was an examination of thermo-energetics with mutant cycle analysis in the fruit fly Kv1 channel (Shaker channel).27) Three critical amino acids were identified for coupling: two charged residues in the S4–S5 linker (Arg-394, Glu-395: numbers correspond to the Shaker sequence) and tryptophan (Tyr-485) in S6. Modeling based on the crystal structure of Kv1.2/2.1 predicts these residues form a complex and suggests the upward motion of S4 causes motion in the complex involving both the S4–S5 linker and the C-terminal end of S6 helix that induces pore opening in the same subunit (Fig. 7a). An all-atom molecular dynamics simulation of the Kv channel response to a membrane potential change also supported this motion of the S4–S5 linker.21) Moreover, recent cryo-EM studies of eukaryotic Nav channels enabled comparison of the structures of the S4–S5 linker in different channel states, which supports the idea of lever lifting motion of the linker upon the motion of S4.28),29)
Of note, in some VGICs the coupling between the VSD and PGD is also chemically regulated. In the KCNQ1 (Kv7.1)/KCNE1 channel complex, which underlies slow outward currents in cardiac muscle, it is known that phosphoinositide (PI) regulates channel activity. Cui’s group showed that PtdIns(4,5)P2 binds to the S4–S5 linker of KCNQ1 to ensure coupling between the VSD and PGD (Fig. 8b),30),31) and a recent cryo-EM structure of KCNQ1 is consistent with that model.32) The TPC1 channel is a multimodal sodium channel activated by both membrane depolarization and binding of PtdIns(3,5)P2, which is most abundant in endosomes/lysosomes, where TPC1 is selectively expressed. An atomic structure of the mammalian TPC1 channel in complex with PtdIns(3,5)P2 showed that PtdIns(3,5)P2 docks near the S4–S5 linker, facing part of S6 close to the cytoplasm and the N-terminus of S3.33)
In Nav, Kv, and Cav channels, the VSD of one subunit is spatially closer to the PGD of a neighboring subunit than the PGD in the same subunit. This raises the natural question, does the VSD of one subunit interact with the PGD of a neighboring subunit? In Kv1.2/2.1, the external end of S1 is situated next to the external side of S5. This interaction probably ensures stabilization of the entire channel structure during dynamic motion of the mobile part (S3–S4) mediating voltage-dependent gating.34) Potential intersubunit interaction between the VSD and PGD has also been observed within the KCNQ1/KCNE1 complex, where a phenylalanine in S4 interacts with a phenylalanine in S5 in the presence of KCNE1. This slows depolarization-activated pore opening without significantly affecting the motion of S4. Amino acid replacement experiments support speculation that such interaction depends on steric hindrance between the two bulky side chains of the aromatic residues.35) In addition, a noncanonical mode of electromechanical coupling involving interfaces between two transmembrane helices, S4 and S5, was recently reported for the Shaker channel.36) Such helix–helix interactions may underlie cooperativity among subunits during gating.
In some VGIC channels, including Kv10,37) Kv11,38) HCN,39) Slo1,10) and Slo2,40) S4 is packed with S5 in the same subunit (i.e., the domain is not swapped), and the S4–S5 linker is much shorter than in authentic VGICs (Fig. 4, left panel). Channels belonging to the domain-nonswapped group have been solved using either X-ray crystallography or cryo-EM. In Kv10.1, coupling between the VSD and PGD does not require physical linkage between the two domains, unlike in domain-swapped VGICs (Fig. 4, Fig. 8). This is illustrated by the fact that upon cutting the channel into two polypeptides at the linker between the VSD and PGD, the channel reassembles to function normally as a Kv channel.41),42) Splitting the linker does not dramatically perturb the channel structure, as abnormal gating is reversed by replacement of a single amino acid near the split site. It has been suggested that slight motion in S4 can compress the S5 helix, triggering the motion in S6, the pore-forming helix.
In Slo1, the large-conductance, Ca2+- and voltage-activated K+ channel, S6 is positioned close to a cytoplasmic ring-like structure, which is connected to the sensing domains for divalent cations (Ca2+ bowl and RCK domain for binding divalent cations). In the HCN channel, S6 is positioned close to the cyclic nucleotide binding domain, which is required for binding cyclic nucleotides. In these domain-nonswapped VGICs, the cytoplasmic region is domain-swapped, in contrast to domain-swapped VGICs. Notably, many domain-nonswapped VGICs are activated not only by a membrane potential change but also by intracellular small molecules, Ca2+ ions (Slo1), Na+ ions (in Slo2.2), cAMP (in HCN), or Ca2+-calmodulin complex (in Kv10). This raises the possibility that the domain-swapped architecture in the cytoplasmic region together with the nondomain swapped architecture of the VSD and PGD may be favorable for dual regulation of S6 motion by both the VSD and the cytoplasmic sensor structure that binds small molecules. However, this idea may be premature, because the TPC channel, which is a domain-swapped VGIC, is regulated by intracellular Ca2+, and the Erg K+ (Kv11) channel is regulated mainly by membrane potential, not through binding small molecules.
To date, the significance of swapping and nonswapping domain interactions between VSD and PGD in the gating mechanisms of VGICs remains unclear.
The VSD was long thought to be unique to VGICs. However, in 2005, we serendipitously discovered an exceptional case in a bioinformatics analysis of the genome of a sea squirt, Ciona intestinalis. We found a VSP has a VSD that is connected not to a PGD but to a cytoplasmic structure with homology to tumor suppressor phosphatase (phosphatase and tensin homolog [PTEN]).43) Using a similar bioinformatics approach, we also identified Hv1, the voltage-gated proton channel, as another example of a VSD lacking a PGD. The VSD of Hv1 functions as both a voltage sensing apparatus and a proton-permeable pore.
3-1. A single VSD couples with the enzyme to change the PI profile in voltage-sensing phosphatase.In all VGICs, multiple VSDs work together to regulate a pore in the center of homologous units (subunits or repeats). In a survey of ion channel-related genes from the whole genome of Ciona intestinalis, several genes were not categorized into previously established groups. Ciona intestinalis (Ci)-VSP is encoded by one such novel gene.43) Ci-VSP shows homology to both the VSD of VGICs and the tumor suppressor PI phosphatase PTEN. Unlike VGICs, VSP lacks a PGD. Within VSP, a single VSD is connected to a cytoplasmic PI phosphatase region with similarity to PTEN.44) The PTEN-like region of VSP contains a phosphatase domain (PD) with a catalytic center and a C2 domain, which associates with the membrane (Fig. 1). Unlike PTEN, VSP lacks a region corresponding to the C-terminal disordered region, which is critical for kinase-dependent regulation of enzyme activity. The linker between the VSD and the PD of VSP, which we call the VSD–PD linker, consists of two parts, a proximal part unique to VSP (i.e., not conserved in PTEN) and a distal part that is conserved between VSP and PTEN. As in PTEN, the distal part of the VSD–PD linker contains numerous positively charged residues able to bind PIs. The mammalian orthologs of Ci-VSP are the PI phosphatases TPTE, TPIP, and PTEN2, which are highly expressed in the testis.45)
The VSP VSD has an architecture similar to other VSDs and consists of four helices, including the signature S4 containing multiple positively charged residues (Fig. 3). A study of the crystal structures of the VSD from Ci-VSP suggests that upon a change in membrane potential, there is a simple upward helical motion of S4 without significant motion change in the other helices.46) Single molecule imaging suggests VSP is a monomer. However, a recent study by the same group showed that VSP proteins can assemble into dimers, depending on their density on the cell surface,47),48) which is consistent with the results of an earlier fluorescence resonance energy transfer (FRET) study of fluorescent proteins.49)
Membrane depolarization activates the enzyme activity of PI phosphatases.50) Unlike with VGICs, quantitative measurement of the readout of VSD action is not straightforward in VSP, where the outcome is enzymatic activity not ion conduction. VSP dephosphorylates PtdIns(3,4,5)P3, PtdIns(3,4)P2, and PtdIns(4,5)P2.51)–53) The robust 5-phosphate phosphatase activity of VSP toward PtdIns(4,5)P2 is in contrast to PTEN, which is highly selective for the 3-phosphate position of PtdIns(3,4,5)P3 and PtdIns(3,4)P2.54) The enzymatic activity of VSP toward PtdIns(4,5)P2 can be monitored through electrophysiological measurement of the activity of coexpressed ion channels that depend on the level of PtdIns(4,5)P2, such as the inward rectifier K+ (Kir) channel (Fig. 5). Alternatively, enzymatic activities can be quantified using fluorescent protein sensors for PIs. Through quantitative measurements of enzyme substrates using these sensors, it was shown that the enzyme activity gradually increases over a wide range of membrane potentials.55) This nature contrasts with shaper voltage-dependent gating of VGICs, which requires activation of multiple VSDs within the channel complex until pore opening (Fig. 6a).

Rapid changes in the Kir current induced by VSP-catalyzed changes in PtdIns(4,5)P2. Shown are representative current traces with voltage steps recorded under two-electrode voltage clamp in a Xenopus oocyte co-expressing an isoform of the Kir3.2 channel (GIRK2d) with the G-protein β and γ subunits and Ci-VSP.50) The G-protein subunits are necessary for activation of the Kir3.2 channel. A hyperpolarizing step from a holding potential of −60 mV to −120 mV activated inward Kir currents. After an episode of depolarization to 100 mV for 1 s, which robustly activated Ci-VSP, PtdIns(4,5)P2 levels are decreased because VSP activity cleaves the 5-phosphate. This leads to a decrease in the Kir current in the second hyperpolarizing step. Endogenous outward currents are elicited during a depolarizing step to 100 mV.

Operation of voltage-sensor containing proteins. In most of voltage-gated ion channels, multiple voltage sensor domains (green) need to take up-state (activate state) until the pore is opened (upper pictures (a)). This is one of the reasons why the voltage dependence of conductance increase in voltage-gated ion channels is very sharp, as represented by voltage-dependent conductance of nerve membranes as first demonstrated by Hodgkin and Huxley. However, it should be noted that some VGICs require only one or two VSDs in the activated state to have the PGD in an open state. Lower pictures (b) show a current model of operation mechanisms of VSP. In contrast with four homologous repeats or subunits in VGICs, single VSD (green) is connected with the cytoplasmic region, which consists of the phosphatase domain (PD)(blue) and C2 domain (light green) in VSP. Upon partial activation of VSD, some phosphatase activity emerges, whereas the enzyme is fully activated upon full activation of VSD. PD has a flexible structure and its association with the membrane is critical for full activation of the enzyme when the VSD is fully activated. This second step requires a special motif, called a “hydrophobic spine” (not drawn in the diagram).60) In both cases, illustration of the motion of S4 follows the classical idea of translation motion for the simplicity of the figure, rather than ideas of transporter model and helical screw model.158)
As has been well established with the VSDs of VGICs, activation of the VSP VSD occurs in several steps that depend on the pairing patterns of the salt bridges between the positive charges on S4 and negative charges on other helices. In fact, gating currents (also called “sensing currents” for charge movements of the VSD of VSP) show some delay in the peak from the initial rise.43) Consistent with this finding, voltage-evoked movements of the VSD, measured from the fluorescence changes of an environment-sensitive fluorophore conjugated to a specific amino acid in the VSD, show more than one component.47) These results indicate the VSP VSD activates in multiple steps.
Given that VSP consists of a single VSD and a downstream enzyme region, a natural question is, does the VSD in a particular activation state induce a specific related level or mode of enzymatic activity? To address this issue, a VSD mutant that assumes a stable intermediate state during VSD activation was studied with zebrafish VSP.56) In this mutant, VSD motion reaches a plateau within a certain voltage range but then increases further at a higher voltage. When enzymatic activity was quantified through measurement of PtdIns(4,5)P2-sensitive Kir channel activity over a wide range of membrane potentials in cell attached patches, the enzymatic activity showed a bi-phasic pattern consistent with the pattern of VSD motion.56) This raises the possibility that distinct states of the VSD are coupled with different modes of enzymatic activity.
In addition to its voltage-dependent regulation, the broader substrate preference (PtdIns(3,4,5)P3, PtdIns(3,4)P2, PtdIns(4,5)P2) is another noteworthy feature of VSP among PI phosphatases. Notably, regulation of PtdIns(3,4)P2 by VSP may be of biological significance, because DF1 fibroblasts transfected with VSP show marked morphological changes, including the development of neurite-like processes, that are dependent on an increase in PtdIns(3,4)P2.57)
Heterologous expression of Ci-VSP with a fluorescent PtdIns(3,4)P2 sensor protein (tandem PH domain containing protein 1 [TAPP1]-derived pleckstrin homology domain (PHD) fused with green fluorescent protein (GFP)) showed that voltage-dependent changes in the PtdIns(3,4)P2 concentration are biphasic: it increases with smaller depolarizations but decreases with larger depolarizations. This result raises the intriguing possibility that the subreaction from PtdIns(3,4,5)P3 to PtdIns(3,4)P2 is more prominent than other subreactions with smaller depolarizations, whereas the subreaction from PtdIns(3,4)P2 to PtdIns4P becomes more prominent with larger depolarizations. This idea was tested in more detail by Isacoff’s group using the versions of Ci-VSP with the VSD trapped in certain states of activation.58) Their results are consistent with a model in which partially activated states of the VSD lead to an enzyme state with a preference for PtdIns(3,4,5)P3, whereas the fully activated state of the VSD leads to an enzyme state with preference for PtdIns(3,4)P2 and PtdIns(4,5)P2. On the other hand, through mathematical modeling, taking into account endogenous enzyme activities, and careful rapid measurements of four PIs upon changes in membrane potential, a simple scheme of common substrate preference among distinct states of the VSD could be fitted to the kinetics of four PI species upon activation of VSP in HEK293T cells.53)
3-2. Mechanisms of coupling between VSD motion and enzyme activity.Analysis of the crystal structure of an isolated cytoplasmic region of Ci-VSP showed distinct protein structures within the substrate binding pocket.52) In particular, a loop structure, called a “gating loop,” was at a key position within the structure of the substrate binding pocket in the PD, raising the possibility that this gating loop regulates the size or profile of the binding pocket and, thus, the on and off of the enzyme. This indicates that local structural changes in PD are responsible for regulating enzymatic activity. However, direct evidence for such dynamic structural rearrangement of the cytoplasmic enzyme region of VSP in live cells has been difficult to be obtained due to the lack of suitable methods. Most tag molecules, such as GFP, cannot be utilized for this purpose because of their bulky structure.
A recent study involving genetic incorporation of an environment-sensitive, fluorescent, unnatural amino acid, 3-(6-acetylnaphthalen-2-ylamino)-2-aminopropanoic acid (Anap) using an orthogonal pair consisting of an amber suppressor tRNA and an engineered amino-acyl-tRNA synthase enabled detection of local structural changes in the enzyme region associated with VSD motion.59) The size of Anap is similar to that of tryptophan. Through genetic incorporation of Anap, several sites within the gating loop (Asp-400, Phe-401, Phe-407, Gln-408) were individually labeled in separate experiments.59) Large changes in Anap fluorescence were detected upon membrane depolarization in all mutants, indicating that the gating loop moves dynamically during coupling with VSD motion. This study also detected changes in the fluorescence of Anap incorporated at sites within the C2 domain, which were believed to be static in PTEN. The speeds of the changes in Anap fluorescence in the gating loop and C2 domain were comparable, indicating that conformational changes for VSD-driven enzymatic activity are not restricted to the PD; they are distributed over the entire enzyme region.
Notably, the intensity of fluorescence from Anap introduced into the C2 domain (e.g., at Lys-555 of Ci-VSP) changed in opposite directions (early decrease and late increase) over the course of a membrane depolarization.59),60) This indicated the enzyme has multiple activated states that are dependent on membrane potential. Recently, a detailed study of a K555Anap mutant revealed that the early and late changes could be isolated through mutagenesis. Introduction of a single amino acid mutation into a novel membrane interface, which we call the hydrophobic spine, enhanced or reduced the late increase in Anap fluorescence. This suggested the second transition (represented by the late increase in Anap fluorescence) depends on the hydrophobic spine, whereas the first transition (represented by the early decrease in Anap fluorescence) does not.60)
Role of the VSD–PD linker in coupling.The 20-amino acid VSD–PD linker is critical for coupling; deletion or mutation of the linker eliminates or reduces the voltage-regulated enzyme activity.43),61)–63) The crystal structures of the VSP enzyme revealed a salt-bridge interaction between a basic amino acid in the VSD–PD linker (R253, R254) and an acidic residue (D400) on the gating loop,52) and mutation of Asp400 markedly weakened the VSD-coupled enzyme activity, suggesting interactions between the VSD–PD linker and the gating loop are critical for the coupling between voltage-induced motion of the VSD and the enzymatic activity of the PD.
Contributing to the complexity of the coupling is that substrate PI can exert an allosteric effect on voltage-regulated enzyme activity. Like the corresponding segment in PTEN, the distal segment of the VSD–PD linker contains multiple basic residues (K252, R253, R254) in a region often called the PI-binding motif (PBM), which likely bind PIs.63) Mutation of the basic residues within this motif markedly weakens coupling between the VSD and enzyme,59),62),63) and depletion of PIs also influences coupling, raising the possibility that by binding to the VSD–PD linker, PIs may facilitate coupling between the VSD and the enzyme. Together with the finding that the VSD–PD linker interacts with the PD via a salt bridge, these results indicate that the VSD–PD linker is a key node for interaction with both the membrane and the PD. Additionally, potential regulation by PI of coupling between the VSD and the downstream effector is reminiscent of the KCNQ1 and TPC1 channels, which require PI binding for efficient coupling between the VSD and PGD.30),31),33) PIs are also known to dock within a space between the VSLD and PGD in TRP channels.64),65) However, direct evidence for physical binding of PI to the VSD–PD linker in VSP is lacking.
Role of the membrane interface in coupling.Given that substrates for VSPs are PIs within membranes, it would be expected that the protein–membrane interface is crucial for the regulation of enzyme activity. Consistent with this idea, the C2 domain, which associates with the membrane, is critical for VSP enzyme activity: deletion of the domain or amino acid replacement within it alters enzyme activity.66),67) A recent molecular dynamics simulation and functional analysis of a heterologous expression system led to the identification of a novel interface between the membrane and the PD, which is critical for the regulation of enzyme activity.60) The molecular dynamics simulation of the PD highlighted two successive conserved hydrophobic residues (L284 and F285 in Ci-VSP) in the PD, which face the membrane. These two hydrophobic residues are followed by a hydrophilic residue, which destabilizes the membrane insertion. This structure appears to provide flexibility to the PD. Mutation of either of the hydrophobic residues markedly changes voltage-dependent enzyme activity. Moreover, the magnitude of the change is graded depending on the hydrophobicity of the side chain of the amino acids. This suggests the hydrophobicity of the hydrophobic spine is critical for enzyme activity. This membrane interface is conserved in PTEN, raising the possibility that the hydrophobic spine is crucial for innate enzyme activity but not for coupling between the VSD and enzyme. However, substitution of an amino acid residue with an aromatic ring in the side chain (e.g., tryptophan) increases voltage-dependent enzyme activity but does not enhance the phosphatase activity of the isolated phosphatase domain, as examined in vitro in a malachite green assay. This indicates that the hydrophobic spine is involved in both the intrinsic enzyme activity and the coupling between the VSD and enzyme. Analysis of fluorescence from K555Anap with a mutation in the hydrophobic spine showed that the voltage-dependent enzyme activity correlates with the second (increasing) component of the fluorescence but not the first (decreasing) component.60) These results lead to three conclusions: (1) the membrane interface is critical for robust enzyme activity; (2) two stepwise transitions underlie enzyme activation upon VSD motion (Fig. 6b); and (3) the second step in enzyme activation, which is mediated by interaction of the hydrophobic spine with the membrane, is required for robust enzyme activity.
3-3. LEGO-blocked VSD of VSP: sophisticated piece for engineering.The VSP VSD can be swapped with another protein domain like a LEGO block to confer voltage sensitivity. Examples of this include VSPTEN, where the VSD of Ci-VSP was fused to the N-terminus of human PTEN.68) VSPTEN exhibits voltage-dependent phosphatase activity toward PtdIns(3,4,5)P3. In another example, the VSD of Ci-VSP was transferred to a virus-derived K+ channel (Kcv).69) With this transfer, voltage-insensitive Kcv became a voltage-dependent K+ channel. In a third example, voltage sensitivity was conferred by transferring the VSP VSD to one or two fluorescent proteins.70)–72) Using this strategy, a variety of gene-encoded voltage indicators (GEVIs) that use the VSP VSD have been designed to visualize electrical activities73),74) and have enabled fluorescence-based detection of single action potentials in CNS neurons.72),75),76) In addition, voltage changes within fine structures, such as dendritic spines, which cannot be accessed using conventional electrophysiological methods, have been studied using ArcLight, a versatile VSP VSD-based GEVI.77),78) The bioluminescent voltage probe LOTUS-V was also designed based on the VSP VSD. Instead of a donor fluorescent protein, LOTUS-V contains a bioluminescent protein, NanoLuc, which is 150 times brighter than firefly luciferase.79) A common problem shared by all fluorescent GEVIs is the phenomenon of photobleaching, which makes long-term monitoring of electrical activities difficult. LOTUS-V does not require illumination and so enables long-term monitoring of cellular electrical activities with a low background signal and little cell damage.73) There are cases in which the VSD of a VGIC was utilized for developing GEVIs.26),80) However, both the expression efficiency and voltage-sensitive signal are low in GEVIs designed based on VSDs from VGICs; consequently, these probes are not practical for monitoring the electrical activities of cells. The better portability of the VSD of VSP than VSDs from VGICs probably reflects the fact that the effector driven by the VSD is a cytoplasmic structure in VSP, not a transmembrane structure like a VGIC.
Within VGICs, positively charged residues on S4 translocate upon changes in membrane potential and change their salt bridge partners (negatively charged residues) on the other helices comprising the VSD, which passes through a constricted hydrophobic barrier in the middle of the electric field. This barrier region faces two water crevices, one from the extracellular side and the other from the intracellular side, and provides a transfer center for positively charged residues in S4. The translocation of the positively charged residues in S4 through this pathway is usually not large enough to be accompanied by ion flow. However, under some conditions it is accompanied by ionic conductance. For instance, Kv12),81) and Nav82) channels containing single amino acid mutations within the helices of their VSDs show state-dependent cation conductance (Fig. 7). When a non-mutated Shaker channel VSD lacking the PGD is expressed heterologously in Xenopus oocytes, proton or monovalent cation conductance emerges.83) These currents are called ω currents or gating pore currents (Fig. 7a) and differ from regular ionic currents through the central pore (which are “α currents”). In Nav channels with mutated VSDs seen in some groups of patients with hypokalemic periodic paralysis, disruption of pH homeostasis of the cytoplasm due to proton-leakage through mutated VSDs likely underlies the pathophysiological changes in skeletal muscle seen in these patients.84)

Examples of ion conductances through VSDs. a. ω-current (gating pore current) through the VSD of a Shaker Kv channel R362C mutant (denoted R1C), in which the first arginine of S4 is replaced with cysteine.81) The PGD is contained in this construct, which was expressed in a Xenopus oocyte. The currents were recorded using patch clamp with 100 mM KCl, 10 mM HEPES, and 1 mM EDTA, pH 7.1 in both the bath and patch pipette solutions. Currents were measured while stepping from a holding potential of −110 mV to test potentials ranging from −200 to +60 mV in 20-mV steps, followed by repolarization to −100 mV. Red traces indicate ω currents evoked by hyperpolarization; black traces are currents through the canonical pore. Some inward currents (black inward traces) appear with small depolarizations because of a negative shift in the I-V curve of the R1C mutant. Transient large outward currents are through the canonical pore. The transient profile of the outward current is due to the fast inactivation of Shaker channels retaining the N-terminal inactivation ball. ω currents (red) do not exhibit such inactivation, which is consistent with the view that ions flow is through a permeation pathway (gating pore of VSD) separate from the canonical pore. b. Voltage-gated proton current through mouse Hv1 heterologously expressed in a HEK293T cell.159) Shown are a family of traces evoked by test pulses stepped from a holding potential of −60 mV to a level ranging from 10 mV to 130 mV in 20-mV increments for 3 s. The bath solution contained (in mM) 180 HEPES, 75 N-Methyl-D-glucamine (NMDG), 1 MgCl2, 1 CaCl2 (pH 6.9). The internal solution contained 183 HEPES, 65 NMDG, 3 MgCl2, 1 EGTA (pH 7.0). pH was adjusted using methanesulfonate. c. Hyperpolarization-activated Ca2+ currents in a HEK293T cell heterologously expressing the VSD from an ascidian CatSper channel subunit, Ci-CatSper3.134) The structure downstream of the VSD including PGD is truncated in this construct. The external solution contained (in mM) 150 NaCl, 2 CaCl2, 10 HEPES (pH 7.4). Internal solution contained 130 CsCl, 1 EGTA, 50 HEPES, pH 7.4. Step pulses were applied from a holding potential of −10 mV to a level ranging from +50 mV to −150 mV in 20-mV increments. The trace at −150 mV is shown in red.
Hv1 (a.k.a. voltage sensor domain only protein, VSOP), a voltage-gated proton channel, has a VSD but lacks a PGD.85)–87) The VSD plays dual voltage sensing and proton permeation roles in Hv1. The salt bridges formed between the three positively charged residues in S4 and negatively charged residues in S1 are critical for proton-selective conduction. The properties of Hv1 currents differ from those of ω currents through VGIC VSDs. Hv1 shows depolarization-induced activation of conductance, whereas most of ω currents are activated by hyperpolarization. In addition, Hv1 tail currents show clear voltage-dependent kinetics (Fig. 7b), whereas the offset of ω currents is not associated with a clear tail current. Amino acid mutation of Hv1 makes the channel conductive upon membrane hyperpolarization, mimicking the ω current without affecting the depolarization-activated outward current.88) This indicated that the mechanism of proton conduction differs from that underlying ω currents through VGIC VSDs.
The most critical characteristic of Hv1 is its high proton selectivity; Hv1 is at least 106 times more conductive for H+ than Na+. Selectivity lower than that does not make sense for a “proton channel,” given that proton concentrations at the pH of normal serum (pH 7.4) corresponds to less than 100 nM, which is about 10−6 times lower than the concentration of Na+ ions in normal serum. The molecular mechanism of proton permeation through Hv1 has been the target of hot debate.89),90) Proposed ideas for proton-selective permeation can be broadly categorized into two models.91) In one model, a water wire is present in the form of a wedge from both sides of the membrane, and water molecules stably sitting in the middle of the constricted area complete the water wire for proton conduction (so called “frozen water” model). In another model, protonation of amino acid side chains underlies shuttling of protons through the channel. D112 in human (h)Hv1 (corresponding to D108 of mouse (m)Hv1) is known to be critical for proton-selective permeation and to form a salt bridge with an asparagine residue in S4. This asparagine residue is thought to be a potentially titratable residue for proton conduction.92) Other signature characteristics of Hv1 are voltage-dependent gating regulated by the pH difference across the membrane, gating inhibition by external zinc ions (as described in a later section),90) and high temperature sensitivity.89),93) Basic properties of voltage-gated proton currents have been explained in earlier reviews.87),89),90),93),94)
Unique among VGICs, Hv1 shows dimeric stoichiometry, with two VSDs within the protein complex. Hv1 has a C-terminal cytoplasmic region with a coiled coil structure95) that has the ability to form a dimer. Coimmunoprecipitation96) and fluorescence labeling96),97) studies showed that Hv1 functions as a dimer within the membrane. Nonetheless, monomeric Hv1 protein lacking the C-terminus still functions as voltage-gated proton channel,96),97) indicating that most of the hallmark features of the channel do not require dimerization.
The crystal structure of mouse Hv1 in a closed state was solved as a chimeric protein (mHv1cc) with the S3–S4 region from Ci-VSP and the coiled coil region from a yeast transcription factor, GCN4.98) Comprising the entire structure are four helices that are slightly oblique and wider at the lower side, like a closed umbrella. S4 runs straight across the membrane, extending its helical structure to the cytoplasmic coiled coil.
4-2. Summary of the biological functions of Hv1.Hv1 is expressed in the immune cells of many species from teleosts to humans, the epithelium of the respiratory system in mammals, and sperm and cancer cells in humans. The Hv1-encoding gene, HVCN1, has also been found in the genomes of marine plankton99) and insects,100) but absent in Caenorhabditis elegans and Drosophila.87),90) Before identification of the Hv1 molecule, voltage-dependent proton conductance was studied extensively with reference to its role in regulating reactive oxygen species (ROS) production in phagocytes.89),101) NADPH oxidase is electrogenic, and the oxidase transfers electrons from cytoplasmic NADPH to oxygen outside membrane to generate ROS. In this reaction, protons are released into the cytoplasm. The electron transfer and proton release cause membrane depolarization and acidification of the cytoplasm. Hv1 channel activity is believed to cancel these effects to maintain NADPH oxidase activity.89) Studies using knockout mice lacking HVCN1 (Hv1-KO) revealed that Hv1 is necessary to maintain ROS production at a high level in neutrophils.102),103) It was also revealed that Hv1 in neutrophils act to inhibit acidification of the cytoplasm and excess depolarization upon NADPH oxidase activation.104) These studies in Hv1-KO mice supported the model that Hv1 maintains NADPH oxidase activity by regulating intracellular pH and membrane potential for ROS production. The regulation of ROS production by Hv1 has also been observed in other immune cells, including T lymphocytes,105) B lymphocytes106) and macrophages.107) However, the detailed mechanisms underlying the regulation of ROS production in these cells remain unclear.
The function of Hv1 in neutrophils appears to be multimodal. Hypochlorous acid (HOCl), a ROS, is generated from hydrogen peroxide (H2O2) by myeloperoxidase (MPO). MPO is released by cells into the extracellular space through exocytosis (degranulation). Hv1-KO neutrophils release more MPO than wild-type neutrophils, resulting in excess HOCl production.108) The enhanced degranulation of MPO-containing granules is suppressed by the NADPH oxidase inhibitor diphenyleneiodonium chloride or the K+ ionophore valinomycin,108) which suppresses membrane depolarization. This suggests that Hv1 inhibits degranulation of MPO-containing granules in neutrophils through the regulation of membrane potential changes, which are induced by NADPH oxidase activation.
Microglia, resident immune cells in the brain, not only protect the brain from invading pathogens, but also clear dead cells and regulate the microenvironment.109) The impact of Hv1 on ROS production in microglia differs from that in other immune cells in that microglia isolated from Hv1-KO mice exhibit increased ROS production.110) The distribution of p67, a NADPH oxidase subunit, and actin dynamics are altered in Hv1-KO microglia, which may underlie the increase in ROS production.110) This idea is consistent with the observation that facilitating actin filament formation using jasplakinolide increases ROS production in wild-type microglia,110) as well as with studies showing that localization of cytosolic NADPH oxidase subunits and ROS production are affected by actin dynamics.111),112) Hv1 probably inhibits ROS production through effects of actin dynamics, although the detailed mechanism remains unknown. These emerging findings indicate that the function of Hv1 in ROS production is more complex than previously thought. Among various cell types, it is likely that Hv1 contributes to cellular function through diverse mechanisms that depend on the physiological or pathophysiological context.
Although the physiological functions of Hv1 have been investigated extensively in Hv1-KO mice, the actions of Hv1 in humans remain unclear. Recently, a potent and highly selective human Hv1 inhibitor, Corza6 (C6), was developed based on the structures of peptide toxins acting on different VGICs.113) Inhibition of Hv1 activity in human neutrophils and sperm using this reagent shed light on the physiological roles of Hv1 in sperm maturation critical for fertilization, so-called capacitation, as well as its enhancement of ROS production in neutrophils.113)
4-3. Twin VSDs work together to regulate proton conduction in Hv1.A remarkable feature of Hv1 channel gating is cooperativity within the dimer; status of one monomer influences the conformational change of the other monomer in the dimer. Based on this cooperativity, dimeric Hv1 shows sharper voltage dependence than the monomer, although it is shallower than that of most of VGICs such as Kv and Nav, which play principal roles in excitable cells.
Cooperative gating of Hv1 was demonstrated by findings obtained in four types of studies employing heterologous expression systems. First, electrophysiological analysis of channel activation and deactivation showed a delay in the activation phase of the dimeric channel, whereas a mono-exponential rise without a delay was seen in the monomeric channel.114),115) In addition, measurements of the limiting slope of activation with small increases in depolarization enabled estimation of the magnitude of the translocated charges per channel, which, in dimeric channels, were double those of monomeric channels.116) Second, analysis of gating kinetics by labeling specific amino acids with an environment-sensitive fluorophore (voltage clamp fluorometry [VCF]) enabled detailed analysis of the activation and deactivation process in VSDs.117) In contrast to measurement of ionic currents, which are the final outcome of a conformational change in an ion channel, VCF permits analysis of conformational changes associated with transitions to partially activated, but nonconductive, states. Experiments using VCF showed that activation occurs in multiple steps and only the last transition of the voltage sensor is coupled to pore opening. Careful comparison of the kinetics of fluorescence changes with the activation kinetics of proton currents led to the idea that there is intimate cooperativity between dimers during the final transition for pore opening.117) Third, a seminal study using the open pore blocker 2-GBI showed that the kinetics of the blockade of two individual pores differ between monomeric and dimeric channels, indicating that the state of one subunit influences the state of the other subunit within the same dimer.118)
How is such coupling between two protomers achieved? For cooperative gating of Hv1, it has been shown that three sites of intersubunit contact (S1, S4, and the coiled coil) between the two protomers are critical (Fig. 8d).119)

Various types of coupling with the VSD among VGICs and voltage sensor domain proteins. a. In domain-swapped VGICs, a complex of the helical linker between S4 and S5 with a part of S6 close to the cytoplasm is critical for transmitting the information of S4 motion to the PGD, leading to pore gating. S4 has a signature alignment of amino acids: several positively charged residues are situated periodically with intervening hydrophobic residues along the helix. b. In domain-swapped, PIP2-sensitive VGICs (e.g., KCNQ1), binding of PI to the S4–S5 linker is critical for coupling. c. In domain-nonswapped VGICs, the VSD couples to the PGD via a linker that is shorter than the linker in domain-swapped VGICs. Both the VSD and PGD interact with cytoplasmic domains that recognize small ligand molecules. Splitting the whole polypeptide within the linker does not eliminate coupling, suggesting helix–helix interaction within the membrane is more important for coupling. d. In Hv1, which lacks a PGD, two VSDs interact through three interfaces: S1–S1, S4–S4, and S4-coiled coil. Dotted arrows indicate the dimer interaction interfaces. Arrows indicate the interaction between S4 and the coiled coil. Within the 3D protein structure, these sites are next to each other when the dimers are in certain conformations. e. VSP lacks a PGD but contains a PTEN-like PI phosphatase. Dephosphorylation of several species of PIs is induced by depolarization-activated motion of the VSD. The upward motion of S4, which harbors several positively charged residues, induces a conformational change in the enzyme. The VSD-phosphatase linker is critical for this coupling, though the structure of the linker remains unknown (dotted box).
The results of cysteine mutagenesis experiments suggested that the external ends of the S1 segments are positioned next to each other within Hv1 dimers through highly efficient crosslinking.120) VCF measurements of VSD motion within Hv1 showed that cooperativity-dependent fluorescence changes were no longer observed after mutation of acidic residues at the external end of S1 in sea squirt Hv1 (CiHv1).117) Open-state coupling between the two protomers, as determined from their sensitivity to an open channel blocker, was also diminished by similar mutations in hHv1.118)
Coiled coil.Biochemical analyses and analysis of the crystal structure of the isolated coiled coil motif showed that this motif in the C-terminal cytoplasmic region of Hv1 has innate activities related to dimer assembly,95),121) and that amino acid mutations within the coiled coil altered the strength of dimer assembly and affect channel gating.121) In addition, electrophysiological characterization of cooperative gating and western blotting analyses of cross-linking showed that S4 forms a continuous helix with the cytoplasmic coiled coil.122) Mutation of the linker region between S4 and the coiled coil modified the extent of cooperative gating, supporting the idea that the S4–coiled coil helical structure provides a core structure for dimer interaction. The continuous helical nature of the S4–coiled coil region was confirmed by two studies of protein structure: an analysis of the crystal structure of the mHv1 chimeric protein mHv1cc98) and an examination of the structure based on electron paramagnetic resonance (EPR) and in silico analyses.123)
S4–S4.Guided by the model of the dimeric structure of mHv1cc,98) the interaction between the two S4s running in parallel through the membrane was closely examined.124) A tryptophan residue in S4 is highly conserved among Hv1 orthologs, and a molecular dynamics simulation predicted that it is positioned at the dimer interface and that the tryptophans in the two protomers are close to one another. Mutation of the tryptophan accelerates deactivation, and it was suggested that S4–S4 interaction occurs during deactivation but not during activation.124) This was consistent with the idea that state transition of the VSD occurs through multiple conformations during activation and deactivation.125)
The crystal structure of mHv1cc in the closed state showed that the S4 segments are close to each other, but the S1 segments are not. Perozo’s group used EPR analysis to study hHv1 and modeled its dimeric structure.123) Notably, the structure remained dimeric even after deletion of a large section of the dimeric coiled coil, which suggests the VSD itself has some capacity for self-assembly as a dimer. Importantly, this structure showed the S1 segments to be close to one another, unlike in the model based on the crystal structure of mHv1cc,98) which was consistent with results from VCF117) and electrophysiological118) studies. It is possible that these two structures represent slightly different states during activation and that a subtle change in the angle across individual helices may cause the neighboring S1 segments to change the state of their interaction.
To summarize, the two Hv1 protomers interact with each other at distinct contact sites on S1, S4, and the coiled-coil domain. The function of each site may vary during cooperative gating, possibly reflecting the particular state during activation/deactivation of the VSD.
4-4. Zinc sensitivity of Hv1: structural basis and biological significance.Another hallmark property of Hv1 is its high Zn2+ sensitivity. External zinc at submicromolar levels inhibits Hv1 gating. As the Zn2+ concentration increases, activation becomes slower, and the activation voltage is shifted to more positive levels, which indicates Zn2+ is not a pore blocker but a gating modifier.89) An early mutagenesis study showed that two histidines (H140 and H193 in hHv1, corresponding to H136 and H189 in mHv1) facing the external side of the channel play important roles in determining Zn2+ sensitivity.86) The crystal structure of mHv1cc showed that two other amino acids also contribute to the Zn2+ sensitivity of mHv1: E115 and D119.98) Although individual mutations of E115 or D119 had no effect on Zn2+ sensitivity, double mutation abolished the sensitivity to Zn2+. A recent attenuated total reflectance-Fourier transform infrared spectroscopy analysis in combination with quantum chemical calculation provided a detailed profile of the Zn2+ coordinating mechanism. Zn2+ is coordinated by N1 (Ni) of the neutral imidazole of histidine, the anionic carboxylate of an acidic residue, and one or two water molecules. The anionic carboxylate of the acidic residue binds Zn2+ in the monodendate mode, and either one or two histidines binds Zn2+.126)
The Zn2+ binding sites are conserved between mouse and human Hv1 orthologs. However, in zebrafish and the African clawed frog, the binding sites are not fully conserved.127) Electrophysiological analyses showed that zebrafish (Dr)Hv1 is resistant to 10 µM Zn2+,127) which strongly inhibits mHv1 currents.85) Of note, the low sensitivity of Dr-Hv1 to Zn2+ appears to be related to the serum Zn2+ concentration, which is around 15 µM in mice and humans,128) but is 10 times higher in zebrafish and clawed frog.127) This suggests the low sensitivity of Dr-Hv1 to Zn2+ is necessary to ensure adequate ROS production by zebrafish neutrophils.
It has been suggested that the Zn2+ sensitivity of Hv1 is physiologically significant in human sperm, which abundantly expresses Hv1.129) Human seminal plasma contains a high concentration of Zn2+ (∼2 mM),130) which diffuses away within the female reproductive tract.131) This supports idea that Hv1 is inhibited by Zn2+ within the testis, but the dilution of Zn2+ within the female reproductive tract leads to Hv1 activation promoting hyperactivation of sperm (capacitation). It has also been suggested that the Zn2+ sensitivity of Hv1 has physiological relevance to host defense by granulocyte-macrophage colony-stimulating factor-treated macrophages.132)
4-5. More examples of ion conductance through the non-mutated VSDs.In addition to Hv1, examples of ion conduction through non-mutated VSDs include CatSper (cation channel of sperm), a Ca2+-permeable channel complex consisting of multiple subunits specifically expressed in spermatozoa. CatSper is the major route for Ca2+ influx into sperm. The α subunit of CatSper shows remarkable similarity to VGICs, and a defect in the α subunit leads to infertility.133) Our recent study showed that a VSLD from the α subunit of sea squirt CatSper3 mediates a slowly activating Ca2+ current upon membrane hyperpolarization in two heterologous expression systems: Xenopus oocytes and HEK293T cells (Fig. 7c).134) Unlike the ω current (or gating-pore current) derived from the Shaker channel VSD,83) this Ca2+ current persists with the expression of the full-length, PGD-containing protein. Moreover, Ca2+ influx was detected upon hyperpolarization to −60 mV, raising the possibility that Ca2+ influx through the VSD could occur within a physiological range of membrane potential. The VSD from mammalian CatSper3 exhibits similar characteristics.135) However, heteromeric assembly of multiple CatSper subunits is known to be essential for normal sperm motility,136) indicating that possible Ca2+ influx through the VSD is not the dominant pathway through the CatSper channel. Whether such conductance through the CatSper VSD actually occurs in spermatocytes remains unknown.
Vriens et al. suggested that TRPM3 has two distinct ion permeation pathways, which are triggered by specific stimuli.137) Ligands such as the neurosteroid pregnenolone sulfate (PS), heat, or nifedipine activate canonical outwardly rectifying currents, whereas hyperpolarization-activated inwardly rectifying currents are induced in the combined presence of PS and clotrimazole. Characteristics of the second current that differ from the canonical current include (1) strong inward rectification, (2) resistance to Ca2+-dependent desensitization, (3) low permeability to Ca2+, (4) low sensitivity to La3+ blockade, and (5) resistance to amino acid mutation and cysteine modification at the central pore. The noncanonical ionic conductance through TRPM3 may underlie drug-induced pain sensation, because clotrimazole potentiates the TRPM3-mediated responses of dorsal root ganglion neurons in vitro and exacerbates PS-induced pain in animal behavior experiments.137) A recent study also showed that the noncanonical conductance is affected by mutation of S4 as well as S1 and S3 of the VSLD, indicating that the noncanonical current is through the VSLD.138) Whether other TRPM subtypes have a similar capacity for ion conduction through the VSD remains to be established.
Proteins containing VSD-like structures continue to emerge. A protein called HVRP1 (Hv1-related protein 1) is abundantly expressed in cerebellar granule neurons and has an amino acid sequence similar to Hv1.139) Its VSLD, which consists of four transmembrane helices, is flanked on the C-terminal side by a coiled coil motif and an extended region that shows no homology to known proteins. Voltage clamp fluorometry showed that the VSLD of HVRP1 senses membrane potential. What kinds of downstream cascades are activated by voltage via HVRP1 remain a mystery. A VSD-like domain has also been found in a sperm-specific isoform of the Na+-H+ exchanger (sNHE or SLC9C1).140) This VSD-like domain consists of four transmembrane segments (S1–S4-like helices) situated downstream of the NHE region. In addition, there is a consensus sequence for a putative cyclic nucleotide-binding domain in the C-terminus. In a recent study of the molecular functions of SpSLC9C1, the sNHE homolog from the sea urchin, Strongylocentrotus purpuratus, the VSLD exhibited gating currents (corresponding to the voltage-dependent charge motion associated with the VSD motion) in a heterologous expression system and conferred voltage-sensitivity to its sodium-proton exchanger activity.141) It will be intriguing to know how the effector domain (NHE domain) in SpSLC9C1 is driven by the C-terminal VSLD.
TRP channels are a large superfamily of cation channels that show significant homology to VGICs. They include TRPV, TRPM, TRPC, TRPA, TRPN, TRPML, and TRPP channels and are activated by diverse chemical and physical stimuli. The protein structure of the transmembrane region of TRPV1, the heat-sensitive TRP channel, is remarkably similar to two features of domain-swapped VGICs.142),143) First, TRPV1 shows domain-swapped positioning of a voltage sensor-like domain (VSLD) and the PGD. Second, the clear α-helical linker present between the VSLD and PGD contains a flex point within S5.143) On the other hand, the cytoplasmic half of the core region contains a number of hydrophobic residues packed into a region corresponding to the water-filling crevice of the VGIC VSD, which can be accessed from the cytoplasm (between S3 and S4). This suggests the VSLD is a rigid structure in TRPV1, which is consistent with the finding that, in yeast, nonbiased random mutagenesis in the S4–S5 linker, but not S1–S4 region, frequently produces a gain-of-function phenotype.144) Given that the S4–S5 linker forms a pocket to bind capsaicin, a TRPV1 activator, the stable nature of the VSLD in TRPV1 ensures that it operates as a stable anchor upon ligand-induced channel activation.142) Notably, S4–S5 is flanked by another conserved helix downstream of the PGD, the TRP motif, which is associated with two N-terminal structures: the ankyrin-repeat and pre-S1 helix. Conservation of the TRP motif among TRP channels suggests a network involving the TRP motif, S4–S5 linker and PGD is central to integration of physical and chemical signals toward pore opening. Liao et al.143) proposed that the similar but distinct structures of VGICs and TRP channels reflect distinct gating modes (voltage versus chemical signals).
Within the structure of TRPV2, the VSLD consists of four helices (S1–S4) containing multiple aromatic residues, suggesting a rigid, immobile structure. A recent study showed that the structure of another TRPV channel, TRPV6, is domain nonswapped.145) It will be interesting in the future to determine the mechanisms by which chemical and physical signals are sensed by TRPV channels and how they differ between domain-nonswapped and domain-swapped channels.
In the TRPML channel, a voltage-sensitive Ca2+-permeable channel in endosomes/lysosomes, PtdIns(3,5)P2 regulates gating. The recently described cryo-EM structure of the TRPML channel revealed a unique cytoplasmic region, termed the mucolipin domain, which connects the VSLD to the PGD. The mucolipin domain consists of cytoplasmic α-helical stretch spanning S2, S3 of the VSLD, and S6 of the PGD. Docking of PtdIns(3,5)P2 to the S2 side of this domain, beneath the VSLD, imposes a pulling force on S6.65) This is in contrast to VGICs, where coupling between the VSD and PGD takes place mainly within the membrane.
As mentioned, in many TRP channels the VSLD is not mobile, but instead appears to operate as a scaffold for PGD gating and is connected to a bulky cytoplasmic region as well as to a ligand docking structure involving the S4–S5 linker. In TRPM channels, S4 underlies voltage-sensitive gating.146) Within the VSLD in TRPM subfamily members, one basic amino acid in S4 is highly conserved at a position corresponding to one of the basic amino acid residues in S4 within VGICs.146) TRPM4 gating is known to be regulated by cytoplasmic Ca2+.147) A recent cryo-EM study of TRPM4 with and without Ca2+ showed that Ca2+ binds within an aqueous pocket in the VSLD and that its binding induces the shift in the position of the side chain of an arginine, which changes its partner amino acid for salt bridging.148) However, whether this positive charge on S4 is located in the focused electric field remains unclear, given that the upper half over the arginine is packed with hydrophobic side chains. The structure of another weakly voltage-sensitive TRPM subfamily member, TRPM8, was also solved using cryo-EM.149) Surprisingly, this structure lacked an obvious S4–S5 linker but showed a pattern of domain-swapping, which is in sharp contrast to the Ca2+-activated K+ channel Slo1. In TRPM8, S4 is long and straight and connects to S5 to form a domain-swapped conformation. For both TRPM4 and TRPM8, it remains unclear whether interactions among helices of the PGD and VSLD are mediated via helix–helix interaction or a pulling motion along the helices.
The cryo-EM structure of the ryanodine receptor (RyR) revealed that the transmembrane segments resemble the structure of the VSD in VGICs, despite little homology at the amino acid sequence level.150) Because RyRs are known to be sensitive to membrane potential, whether this voltage dependence is derived from the VSD-like structure in RyRs is an important issue. A structure with some similarity to the VSD has also been found in the TRIC channel, a cation channel in the ER membrane.151) TRIC channels are trimeric with a pore in each individual protomer. One of the transmembrane helices, M4, contains three conserved positively charged residues facing one side of the protein,151) which is reminiscent of the pattern in S4 of the VSD. Given that TRIC channels show clear voltage-dependence, whether the positive charges in M4 contribute to voltage sensing is an intriguing question.
In its most basic form, the VSD operates as an ion channel by itself. In Hv1, two protomers interact with one another to exhibit cooperative gating. In VSP, one VSD regulates an intrinsic enzyme. In VGICs, one channel protein complex consists of four VSDs and four PGDs. At a higher level, Cav1.1 interacts with the RyR. In that case, the stoichiometry of the Cav1.1-RyR complex is 4:1, with a single Cav1.1 binding to one subunit within the tetrameric RyR.152) In other words, a complex consisting of one RyR with four Cav1.1 channels contains 16 VSDs. In peripheral neurons, Cav2.2 directly interacts with SNARE complex, and a voltage-dependent conformational change in Cav2.2 regulates SNARE complex without requiring Ca2+ influx. This was identified as the phenomenon underlying calcium independent, voltage dependent secretion.153)
Four-repeat type VGICs, including Cav and eukaryotic Nav, have long been thought to operate as monomers. No evidence has yet been presented for cooperativity mediated through physical contact among clustered, multiple channels, except perhaps the pyramidal shape of clusters of Cav1.1-RyR complexes in skeletal muscle. On the other hand, Cav1.3 and Nav1 were recently reported to exhibit cooperativity through physical interaction among individual channels on cell membranes.154),155) This suggests VGICs may transmit voltage-dependent information through interaction with binding partners (homologous or heterologous interactions) or by forming higher-order signaling complexes.
In this review, we described VSD functions in VGICs and in two VSD proteins that lack a PGD. VGICs employ multiple mechanisms for coupling to the VSD, including interaction among transmembrane helices and mechanical motion of the helix linker between the two domains (Fig. 8a, 8b, 8c). Unraveling the mechanisms of VSD-PGD coupling in VGICs will be important for understanding the pathophysiology of diseases caused by mutation or alteration of VGICs as well as for drug development. In VSP, the VSD is coupled to an enzyme, and the linker and the membrane interfaces are critical for this coupling (Fig. 6). In Hv1, the VSD itself operates as both a channel pore and voltage sensor. Two VSDs behave as a single unit through tight interaction that is dependent on multiple interfaces, including transmembrane helices and the cytoplasmic coiled coil (Fig. 8d).
Comparison between VGICs and the two VSD proteins lacking PGDs provides a unique opportunity to gain insights into the common mechanisms of VSD dynamics and VSD-effector coupling. For example, the helical linker downstream of S4 is critical for downstream signaling in VGICs, VSP and Hv1. The mechanisms operating in VSD-containing proteins may also be shared by other sensors, such as light-sensitive proteins. In addition, these findings will be useful to strategically devise better molecular tools for imaging membrane voltage or for manipulating cell signals.
Finally, we stress that there is a gap between the biophysical mechanisms of VSD-effector coupling and the physiological functions of VSD-containing proteins. For example, little is known about the physiological function of VSP in any animal species. VSP is known to be expressed in sperm and epithelium,61) but the patterns of membrane potential change in VSP-expressing cells remain largely unknown, in part due to the limited access of conventional methods of electrophysiology. VSP exhibits robust and rapid changes in enzymatic activity when it is expressed heterologously. For this reason, VSP has often been used as a molecular tool to alter PIs.61) From the standpoint of membrane physiology, however, it is surprising that the enzymatic activity of VSP continues to increase at membrane potentials over 60 mV, levels that cannot be attained in a physiological context in most cell types. Moreover, the molecular properties of mammalian orthologs of VSP remain unknown, in large part because functional characterization of full-length mammalian VSP orthologs in heterologous expression systems has been unsuccessful. In Hv1, dimer cooperativity in voltage-dependent gating has been extensively studied for its unique nature as an ion channel,117)–119),121),122),156) but its actual physiological significance remains unclear. To understand how the molecular design of these VSD proteins is optimized for their physiological function, more coherence between studies of the proteins themselves (such as biophysical characterization in heterologous expression systems or structural studies of their molecular nature) and elucidation of their molecular behavior within native cells in physiological contexts will be necessary.
We would like to thank Dr. Jianmin Cui for helpful comments and careful reading of the manuscript. We thank Ms. Yuka Jinno, Dr. Akira Kawanabe, and Dr. Souhei Sakata for help with the artwork of the figures, and lab members and team members of the Atsushi Nakagawa CREST project (JPMJCR14M3) for helpful discussions. We would also like to thank our many collaborators for all their contributions over the years, which have enabled us to write this review. This work is supported by Grants-in-Aid from the Japan Society for the Promotion of Science (JSPS) (JP21229003 to Ya. O. and Yo. O., JP25253016 to Ya. O., JP16H02617 to Ya. O. and Yo. O., 15K08175 to Yo. O.), Ministry of Education, Culture, Sports, Science, and Technology (MEXT) (JP24111529, JP26111712, JP15H05901 to Ya. O.), and Core Research for Evolutional Science and Technology (CREST, JST) (JPMJCR14M3).

Yasushi Okamura was born in Tokyo in 1960 and graduated from the School of Medicine, the University of Tokyo and obtained his license as a medical doctor in 1985. He went to the Graduate School of Medicine, the University of Tokyo and received his Ph.D. degree in 1989.
After working as a postdoctoral fellow at the State University of New York at Stony Brook between 1989 and 1990 under the supervision by Dr. Gail Mandel (currently a researcher at the Howard Hughes Medical Institute and Professor at the Vollum Institute, Oregon Health and Science University), he was a lecturer at the Department of Neurobiology, Institute for Brain Research, Faculty of Medicine, the University of Tokyo until 1995. He was senior researcher and group leader of the Biomolecular Engineering Department, National Institute of Bioscience and Human-Technology, Agency of Industrial Sciences and Technology in Japan between 1995 and 2001. In 2001, he was appointed as a full professor at the Okazaki Institute for Integrative Bioscience in Okazaki. Since 2008, he has been a professor at the Graduate School of Medicine, Osaka University. For a long time, he has been working on ion channels, in particular their structure–function relationships and regulation of expression. He pioneered a new research field of voltage-evoked cell signals by his discovery of a novel class of membrane proteins that have voltage sensor domains but lack an authentic pore domain.

Yoshifumi Okochi was born in Aichi in 1973 and graduated from the School of Agriculture, Hokkaido University. He went to the Graduate School of Science, Nagoya University and received his Ph.D. degree in 2005. He worked as a postdoctoral fellow at the Okazaki Institute for Integrative Bioscience in Okazaki from 2005 to 2008 under the supervision by Dr. Yasushi Okamura. Since 2008, he has been an assistant professor at Graduate School of Medicine, Osaka University. He has been working on ion channel functions at both the cellular and whole body levels.