Tyrosinase Inhibitors from Natural and Synthetic Sources as Skin-lightening Agents
2019 Volume 7 Pages 41-58
Details
2019 Volume 7 Pages 41-58
Melanin, a major pigment in mammalian skin, is known to protect the skin against harmful effects of ultraviolet (UV) irradiation, oxidative stress, and DNA damage. The accumulation or over production of melanin can cause esthetic problem as well as serious diseases related to hyperpigmentation. Tyrosinase, is a copper-containing enzyme which catalyses two rate–limiting reactions in melanogenesis: the hydroxylation of monophenols to o-diphenols, and the oxidation of o-diphenols to o-quinones. Therefore, inhibition of tyrosinase, is the prime target for researchers to regulate melanin production. Tyrosinase inhibitors with high efficacy and less adverse side effects, have huge demand in cosmetic and medicinal industries due to their preventive effect on pigmentation disorders as well as skin-whitening effect. In this review, we focus on the recent advances of tyrosinase inhibitors from all sources, including synthesized compounds, natural products, virtual screening and structure-based molecular docking studies; by categorized into two parts, mushroom and human tyrosinase inhibitors.
Cosmetic skin concerns have a major impact on the quality of life of an individual (Salsberg et al., 2016). Skin lightening is defined as the procedure of using natural or synthetic products to lighten the skin tone, or provide an even skin complexion by reducing the melanin content in the skin and thus skin lightening products helps people to treat their skin problems such as acne scars, age spots, freckles, or discoloration. Desire for flawless and radiant skin, the demand for skin lightening products is not only limited to women but also a significant rise from men as well, particularly in countries of Asia-Pacific. Skin whitening products are largely popular in Asian countries including China, India, Japan, Korea, and Indonesia. As projected by the latest research report published by Fact.MR, the global market for skin lightening products are estimated to account for over US$ 23,000 million by 2022-end (www.factmr.com).
Melanin, a major pigment in mammalian skin, is produced form melanocytes by the enzymatic oxidation of tyrosine. It is known to protect the skin against harmful effects of ultraviolet (UV) irradiation, oxidative stress, and DNA damage. Moreover, it is also thought to play an important role in the prevention of skin cancer by protecting cells from UV rays. However, it is believed that melanin is also a reason for sunburns and mottle; therefore, compounds inhibiting melanin synthesis are expected to have cosmetic applications as whitening agents (Yamauchi and Mitsunaga, 2016; Baek et al., 2015). Melanin is synthesized in melanosomes and subsequently transferred to the surrounding epidermal keratinocytes (Ando et al., 2007). Melanogenesis is regulated by several melanocyte-specific enzymes such as tyrosinase, tyrosinase-related protein 1 (TRP1), and tyrosinase-related protein 2 (TRP2) (Kobayashi et al., 1994; Yokoyama et al., 1994; Pawelek and Korner, 1982).
Tyrosinase, a binuclear copper enzyme, is a key regulatory enzyme of melanogenesis. Tyrosinase catalyzes the rate limiting reactions of melanin synthesis: the hydroxylation of tyrosine and the oxidation of 3,4-dihydroxyphenylalanine (L-DOPA) to o-dopaquinone. The color of skin is determined by the type and amount of melanin synthesized, and its distribution in the surrounding keratinocytes. Inhibition of tyrosinase with high efficacy and less adverse side effects, has been a long standing challenge in dermatological and cosmetological sciences (Briganti et al., 2003).
Tyrosinase (EC 1.14.18.1), also known as phenol oxidase, is a copper-containing enzyme which catalyses a similar phenol oxidation reaction to peroxidase. This enzyme is widely distributed in nature. Tyrosinase catalyses two main reactions. The first reaction is the hydroxylation of monophenols leading to o-diphenols, often known as monophenolase or cresolase. The second reaction is the oxidation of o-diphenols to o-quinones, often referred to as o-diphenolase or catecholase. In the both of these oxidation reactions, oxygen is used as an oxidant.
Bourquelot and Bertrand in 1895, first isolated tyrosinase from mushroom. After that, tyrosinase has been isolated and purified from a number of bacteria, fungi, plant and animal sources (Table 1). The molecular weight of tyrosinase ranges from 13.4 kDa to 128 kDa depending on the sources (Duckworth and Coleman, 1970; Solomon et al., 1996). The structure of tyrosinase contains three domains: the central domain, the N‐terminal domain, and the C‐terminal domain (Van Gelder et al., 1997). The central domain composed of six conserved histidine residues, coordinating the two copper ions (CuA and CuB) (Figure 1). The spectroscopic properties of the copper ions are similar in all type‐3 copper proteins suggesting that the differences in activity may be attributed to the architecture and substrate accessibility of the different members (Decker et al., 2006). Matoba et al. determined the three-dimensional structure of Streptomyces castaneoglobisporus tyrosinase in complex with caddie and suggested that two copper ions are transported to the tyrosinase catalytic center with the assistance of caddie protein which act as a metallochaperone.
| Scientific name/Common name | Number of Amino acids | Molecular weight (kDa) |
| Homo sapiens (Human) |
529 | 60.4 |
| Mus musculus (Domestic mouse) |
533 | 60.6 |
| Rana nigromaculata (Black spotted frog) |
60.6 | 60.1 |
| Oryzias latipes (Rice fish) |
540 | 61.3 |
| Agaricus bisporus (Button mushroom) |
556 | 64 |
| Neurospora crassa (Filamentous fungus) |
685 | 75.9 |
| Lycopersicon esclentum (Tomato) |
585–630 | 70.5 |
| Streptomyces antibioticus (Bacteria Gram+) |
272 | 30.5 |
| Rhizobium melitot (Bacteria Gram-) |
494 | 54 |
| Ipomea batatas (Sweet potato) |
496 | 39 |

Active site of Agaricus bisporus tyrosinase (modified from Ramsden et al., 2014). Cu, O and His are Cupper ion, Oxygen and Histidine.
[From 3D structure, we have drawn 2D and ignored the binding of other amino acids and atoms except Cu, O and His]
During catalysis, the type-3 copper center of tyrosinase adopts three redox forms: deoxytyrosinase, oxytyrosinase and mettyrosinase form (Solomon et al., 1996). The deoxy form (Cu(I)-Cu(I)) is a reduced species, which binds dioxygen to yield the oxy form. In the oxy form, molecular oxygen binds in the form of peroxide (Cu(II)-O22−-Cu(II)), and activates it. The met form (Cu(II)-Cu(II)) is recognized as the resting enzymatic form. In these three redox forms of tyrosinase(Figure 2), the oxy form can catalyze both monooxygenase and oxidase reactions, whereas the met form lacks monooxygenase activity.

Catalytic cycles of the hydroxylation of monophenol and oxidation of o-diphenol to o-quinone by tyrosinase (modified from Chang, 2009). E, Em, and Ed are the oxytyrosinase, mettyrosinase and deoxytyrosinase respectively. EoD, EoM, EmM and EmD are Eo-Diphenol, Eo-Monophenol, Em-Monophenol and Em- Diphenol complexes, respectively.
[We have added the schematic drawing of different redox form of tyrosinase - oxytyrosinase, mettyrosinase and deoxytyrosinase and the abbreviated symbol are different]
The above considerations guided to the molecular mechanism for the monophenolase and diphenolase activity of tyrosinase (Figure 2). Several study reported the mechanism for the monophenolase activity of tyrosinase (Wilcox et al., 1985; Burton, 1994). In the monophenolase cycle, the monophenol can react only with the oxy form and be catalyzed to a coordinated o-diphenol, which is oxidized to the o-quinone, resulting in a deoxy form ready for further dioxygen binding. The binding of molecular oxygen to deoxytyrosinase can regenerated oxytyrosinase. In the diphenolase cycle, if only o-diphenol is present, both the oxy and met forms react with o-diphenol to yeild the o-quinone. The met form of the enzyme is generated through the binding of o-diphenol to the oxy form and oxidized to o-quinone. The met form transforms another o-diphenol molecule into o-quinone and is reduced to the bicuprous deoxy form. The met form is considered as an important target for the discovery of tyrosinase inhibitors.
Melanin biosynthesis takes place within membrane-bound organelles called melanosomes in melanocytes. Melanosomes are the synthesis, storage and transportation site for melanin pigment in the cells. Skin color is a function of the size, number and distribution of melanosomes. Melanocytes are distributed in the epidermis, hair follicles, the inner ear and the eye. Melanocytes in mammals produce two chemically distinct types of melanin, black to brown eumelanin and yellow to reddish-brown pheomelanin. Most natural melanin pigments contain eumelanin and pheomelanin and are termed 'mixed' melanins. The biosynthetic pathway for melanin formation in various life forms has been elucidated by Raper, 1928; Mason, 1948; Schallreuter et al., 2008 (Figure 3). Melanogenesis is initiated by the tyrosinase (TYR)-catalyzed oxidation of tyrosine to dopaquinone via the intermediate, 3,4-dihydroxyphenylalanine. This oxidation step is the rate-limiting step in melanin synthesis because the remainder of the reaction sequence can proceed spontaneously at a physiological pH value (Halaban et al., 2002). Eumelanin are formed through a series of oxidation reactions from dihydroxyindole (DHI) and dihydroxyindole-2-carboxylic acid (DHICA), which are the reaction products from dopachrome. The second enzyme in the pathway, tyrosinase-related protein 2 (TYRP-2; dopachrome tautomerase), enables the rapid conversion of dopaquinone to dopachrome, and then to 5,6-dihydroxyindole (DHI) or indole 5,6-quinone 2-carboxylic acid (DHICA). TYRP-1 (DHICA oxidase) then catalyzes the oxidation of DHICA to produce eumelanin. In the presence of cysteine or glutathione, dopaquinone is converted to cysteinyldopa or glutathionyldopa and finally can produce pheomelanin. Although three enzymes (TYR, TRP-1 and TRP-2) are involved in the melanogenesis pathway, only TYR is exclusively necessary as rate limiting catalyst for melanogenesis.

Biosynthetic pathway of melanin (Yamauchi and Mitsunaga, 2016). Abbreviations: TYR, tyrosinase; l-Dopa, l-3,4-dihydroxyphenylalanine; DHICA, 5,6-dihydroxyindole-2 carboxylic acid; DHI, 5,6-dihydroxyindole
Cutaneous pigmentation is under complex genetic control regulated by more than 150 alleles spread over 90 loci. Protein products of these loci acting as enzymes, structural proteins, transcriptional regulators, transporters, receptors, and growth factors have a wide array of functions and cellular targets (Silvers, 1979). UV exposure or other environmental stimulations, can triggered melanogenesis via different homones or cytokines. Figure 4 shows the most commonly known signal pathways involved in the regulation of melanogenesis. All signaling pathways involve MITF, a master regulator of melanogenesis, which upregulates melanogenesis enzymes TYR, TRP-1 and TRP-2 via binding to the M-box motif in their promoter regions . The transcription factor MITF, which has been reported to activate more than 25 genes in pigment cells, has emerged as an essential regulator not only for melanocyte development, proliferation and survival, but also for the expression of enzymes and structural proteins ensuring the production of melanin (Vachtenheim and Borovanský, 2010). The up-regulation or down-regulation of MITF activity activates or suppress the expression of the melanogenesis-related enzymes, thus stimulating or inhibit melanogenesis.

Common signal pathways involved in the regulation of melanogenesis (Pillaiyar et al., 2017b).
Alpha melanocyte-stimulating hormone (α-MSH) regulates melanogenesis via cyclic adenosine monophosphate/protein kinase A (cAMP/PKA) mediated pathway (Busca and Ballotti, 2000). When α-MSH binds to its receptor, melanocortin 1 receptor (MC1R), on melanocyte membrane activates adenylate cyclase (AC) to produce cAMP, which activates protein kinase A (PKA) by phosphorylation. Phosporylated PKA phosphorylates cAMP-response element binding (CREB) protein in the nucleus and in turn, upregulates MITF (Gonzalez and Montminy, 1989). Ultimately, MITF efficiently activates the melanogenesis-related enzymes and stimulates melanogenesis. β-MSH and adrenocorticotropic hormone (ACTH), also stimulate melanogenesis via the same pathway. cAMP also affects the post-translational modification of MITF through inhibition of phosphatidylinositol 3-kinase (PI3K), which results in reduction of Akt phosphorylation and thereby stimulating glycogen synthase kinase 3β (GSKβ) activity by reducing p-GSKβ levels. The activated GSKβ, phosphorylates MITF and leading to stimulation of melanogenesis by upregution of MITF (Saha et al., 2006).
Wnt signal pathway also targeting the gene expression of MITF by increasing the level of intracellular β-catenin. Activation of the Wnt pathway through binding to a G-protein-coupled receptor (called Frizzled), negatively regulates GSK-3β, leading to the accumulation of cytoplasmic β-catenin, which translocates to nuclei and forms a complex with both T-cell factor (TCF) and lymphocyte enhancer factor-1 (LEF) to up-regulate expression of the MITF gene (Takeda et al., 2000; Bellei et al., 2012) , and thus stimulates melanogenesis. NO signaling also stimulate melanogenesis by activating guanylyl cyclase (GC) which induces the production of cGMP, and, consequently, MITF expression and melanogenesis (Roméro-Graillet et al., 1997).
In contrast to other signal pathways, the extracellular signal-regulated kinase (ERK) or JNK pathway regulates melanogenesis via the degradation of the MITF protein (Kim et al., 2006). ERK activation phosphorylates MITF, which is followed by MITF ubiquitination and proteasome-mediated degradation (Kim et al., 2003a). Therefore, activation of the ERK pathway would inhibit melanogenesis due to the down-regulation of the MITF activity.
Regulation of melanogenesis in mammals is controlled at different levels, started from genetic level during embryo development, at the cellular level via the controlling formation of melanosomes, at the subcellular level where the gene expression encoded by the melanogenesis-related enzymes, including tyrosinase, TRP1 and TRP2 and finally, when transport of melanin pigment from melanocytes to keratinocytes where it produce color. Melanogenesis inhibitors as skin-whitening agents are very important to the cosmetic industry. Skin whitening agents work by depigmenting activity. Depigmentation can be achieved by several mechanisms but one of the prime target by regulating activity of tyrosinase. Tyrosinase is the rate-limiting enzyme of melanin production and, accordingly, is the most prominent target for inhibiting hyperpigmentation. Inhibition of tyrosinase with high efficacy and less adverse side effects, has been a long standing challenge in dermatological and cosmetological sciences. In this review, we focus on the recent advances of tyrosinase inhibitors from all sources, including synthesized compounds, natural products, virtual screening and structure-based molecular docking studies.
A huge number of tyrosinase inhibitors have been identified from both natural and synthetic sources. The formation of dopachrome in the presence of tyrosine or dopa as the substrate, is assessed to examine many putative inhibitors. Chang (2009) discussed several ways to achieve anti-tyrosinase activity. It can be done by the reducing agents such as ascorbic acid, which can reduce o-dopaquinone to dopa or by the o-dopaquinone scavenger such as thio-containing compounds, which can react with dopaquinone to form colorless products. The alternative enzyme substrates such as some phenolic compounds can inhibit tyosinase activity through preventing the dopachrome formation. Some nonspecific enzyme inactivators such as acids or bases can also represent anti-tyrosinase activity by denaturing the enzyme. The specific tyrosinase inactivators such as mechanism-based inhibitors, which can inhibit tyrosinase activity by inducing the enzyme catalyzing ‘suicide reaction’. And finally the most significant, specific tyrosinase inhibitors that reversibly bind to tyrosinase and reduce its catalytic capacity. Among them, only specific tyrosinase inactivators and inhibitors are called as ‘true inhibitors’ because they actually bind to the enzyme and inhibit its activity. Based on inhibition mechanism, true inhibitors are classified into four types: competitive, un-competitive, mixed type and non-competitive inhibitors.
Tyrosinase, the key regulatory enzyme of melanogenesis, is the prime target to inhibit melanin production. Commercially available skin lighting agents are mostly tyrosinase inhibitors. Many tyrosinase inhibitors have been used as skin-whitening agents (Figure 5) but most of them contains specific drawbacks (Pillaiyar et al., 2017a). Hydroquinone, the most frequently prescribed ingredients for skin-lighting agent have adverse effect such as skin irritation and burning, mutagenic to mammalian cells and cytotoxic to melanocytes (Parvez et al., 2006; Curto et al., 1999). Kojic acid and arbutin also show poor efficacy in vivo, low formulation stability and poor skin penetration (Hermanns et al., 2000). The use of kojic acid in skin treatment is limited due to its carcinogenicity (Fuyuno, 2004). Ascorbic acid is liable to degradation (Spínola et al., 2013) and the bioavailability of ellagic acid is poor (Arulmozhi et al., 2013). Thus, it is in great need of developing new tyrosinase inhibitors from different sources with high efficacy and less adverse side effect. Here, the tyrosinase inhibitors were mainly categorized into two parts, mushroom and human tyrosinase inhibitors and their recent advances in both natural and synthetic sources are going to be discussed.

Chemical structure of standard tyrosinase inhibitors as skin lightening agents
Kojic acid, a fungal metabolite used as skin-whitening agent is the most studied inhibitor of tyrosinase. Kojic acid can chelate copper at the active site of the enzyme and show competitive inhibitory effect. The popular whitening agent kojic acid together with arbutin, hydroquinone or tropolone used as a positive control to screen for finding inhibitors. In most of the study, the tyrosinase activity is expressed as the half-maximal inhibitory concentration (IC50), which is the concentration of the samples producing 50% inhibition. B16F10 murine melanoma cells also frequently used for in vitro screening as they share most of the melanogenic mechanisms of normal human melanocytes and they are relatively easy to culture in vitro. Recently, for in vitro screening, reconstituted 3-dimensional (3D) human skin equivalent model also used. Zebrafish used as an in vivo model to evaluate anti-pigmentary effect as their genetic make-up and organ system are similar to human. Moreover, it’s easy to maintain and high efficacy in drug penetration through skin.
There is an ongoing effort to search for tyrosinase inhibitors from natural sources particularly from plants as they are a rich source of bioactive chemicals and are mostly free from harmful side effects. A number of research have been dedicated to identify tyrosinase inhibitors from plants, fungal metabolites and marine algae. Polyphenols are widely distributed in nature and are the largest groups in tyrosinase inhibitors. Best-studied polyphenols are flavonoids, that may be subdivided into seven major groups, including flavones, flavonols, flavanones, flavanols, isoflavonoids, chalcones, and catechin. In addition to flavonoids, stilbenes and coumarin derivatives; long-chain lipids and steroids; benzaldehyde and benzoate derivatives also identified as tyrosinase inhibitors. A large number of compounds have been identified from the natural products and investigated for mushroom tyrosinase inhibitory activity; these compounds differ from one another in the potency and type of inhibition imposed on the enzyme. Here listed some tyrosinase inhibitors (Figure 6) identified from natural sources whose potency is equal to or better than the kojic acid, a well-known positive control for tyosinase inhibitor.

Chemical structure of some potent tyrosinase inhibitors from natural sources
The availability of efficient technology to rationally design efficient synthetic analogs, many synthetic tyrosinase inhibitors with the novel structural moiety together with derivatives of natural compounds have been prepared. Lee et al. (2016) reviewed on natural, semisynthetic and synthetic tyrosinase inhibitors, and mentioned that the development of many synthetic tyrosinase inhibitors (such as N-hydroxy-N′-phenylthiourea, N-cyclopentyl-N-nitrosohydroxylamine, sildenafil methyl ether, 2,5-disubstituted-oxadiazoles, oxazolones, tetraketones, 4,4′-dihydroxybiphenyl, S-phenyl N-aryl S-alkylthiocarbamates, triazolothiadiazoles, benzaldehyde thiosemicarbazones, 4-hydroxybenzaldehyde derivatives, bis-salicylaldehydes, phenylurenyl chalcones, polyphenolic curcumin derivatives, bis-benzyl glycosides, rhodanine derivatives etc.) have been reported, and the properties of those synthetic inhibitors were thoroughly reviewed by Loizzo et al.(2012) & Kim and Uyama (2005). Figure 7, showed some selective tyrosinase inhibitors from synthetic sources whose potency is better than kojic acid in terms of IC50 value.

Chemical structure of some potent synthetic tyrosinase inhibitors
Carvacrol is a naturally occurring monoterpene phenol, having odoriferous and antimicrobial activities, used as meat preservatives or flavoring agents in the food industry. Ashraf et al. (2017) synthesized carvacrol derivatives 1-6 (benzoic acid substituted) and 7-10 (cinnamic acid substituted), with the aim to possess potent tyrosinase inhibitory activity (Figure 8). Among the derivativecompounds, 9(2-[2-methyl-5-(propan-2-yl)phenoxy]-2-oxoethyl(2E)-3-(2,4dihydroxyphenyl)prop-2-enoate) found to possess strongest anti-tyrosinase activity (IC50 0.0167μM) compare with standard kojic acid (IC50 16.69μM). The derivative compounds 3 and 8 also showed good tyrosinase inhibitory activity with IC50 6.7 μM and 6.5 μM respectively. The kinetic analysis revealed that compounds 3 and 8 showed mixed-type inhibition while 9 is a non-competitive inhibitor having Ki values 19μM, 10μM, and 0.05μM respectively. Docking studies showed that compound 9 have maximum binding affinity against mushroom tyrosinase with binding energy value (-7.90 kcal/mol) as compared to others. From structure–activity relationship, the hydroxy substituted derivatives showed better tyrosinase inhibitory activity. The key factor of inhibitory activity is the substitution pattern of hydroxyl groups at phenyl ring. The derivatives with hydroxy substituted cinnamic acid residue possess greater tyrosinase inhibitory potential as compared to benzoic acids. The compound 9 exhibited excellent tyrosinase inhibitory activity (IC50 0.0167μM) bearing 2,4-dihydroxy substituted cinnamic acid residue. The study proposed that the hydroxy substitution pattern on phenyl ring in case of compound 9 impedes the molecule to interact well with the active sites of enzyme.

(A) Chemical structure of carvacrol derivatives 1-10
(B) Tyrosinase inhibitory activity of carvacrol derivatives 1-10
Polyphenol compounds containing sulfur atom can interact with cupper ion in the active side of mushroom tyrosinase and inhibits the activity. Chen et al. (2015) extracted bis(4-hydroxybenzyl)sulfide, from Gastrodia elata and found outstanding inhibitory potency against mushroom tyrosinase (IC50 = 0.53 µM, Ki = 58 ± 6 nM), which is more effective than β-arbutin, kojic acid, short peptides and other known natural compounds. Bis(4-hydroxybenzyl)sulfide (Figure 9A) may have function as a copper chelator to abolish the tyrosinase activity and a strong competitive inhibitor of mushroom tyrosinase. In docking study, the sulfur atom of bis(4-hydroxybenzyl)sulfide makes close contacts with the copper ions of tyrosinase. In addition, two hydrogen bonds were observed between the hydroxyl groups of bis(4-hydroxybenzyl)sulfide and Asn260 and His224, respectively. The side chains of residues Glu256, Phe90, Val238, and Phe264 show tightly hydrophobic contacts and/or π-π interactions with the molecule. Bis(4-hydroxybenzyl)sulfide adapts the same orientation as interacting with mushroom tyrosinase to interact with human tyrosinase and more specific to mushroom instead of human tyrosinase. The in vitro and in vivo assay, reveals that this compound effectively reduces melanogenesis without any adverse side effects and is free of discernable cytotoxicity in mice.
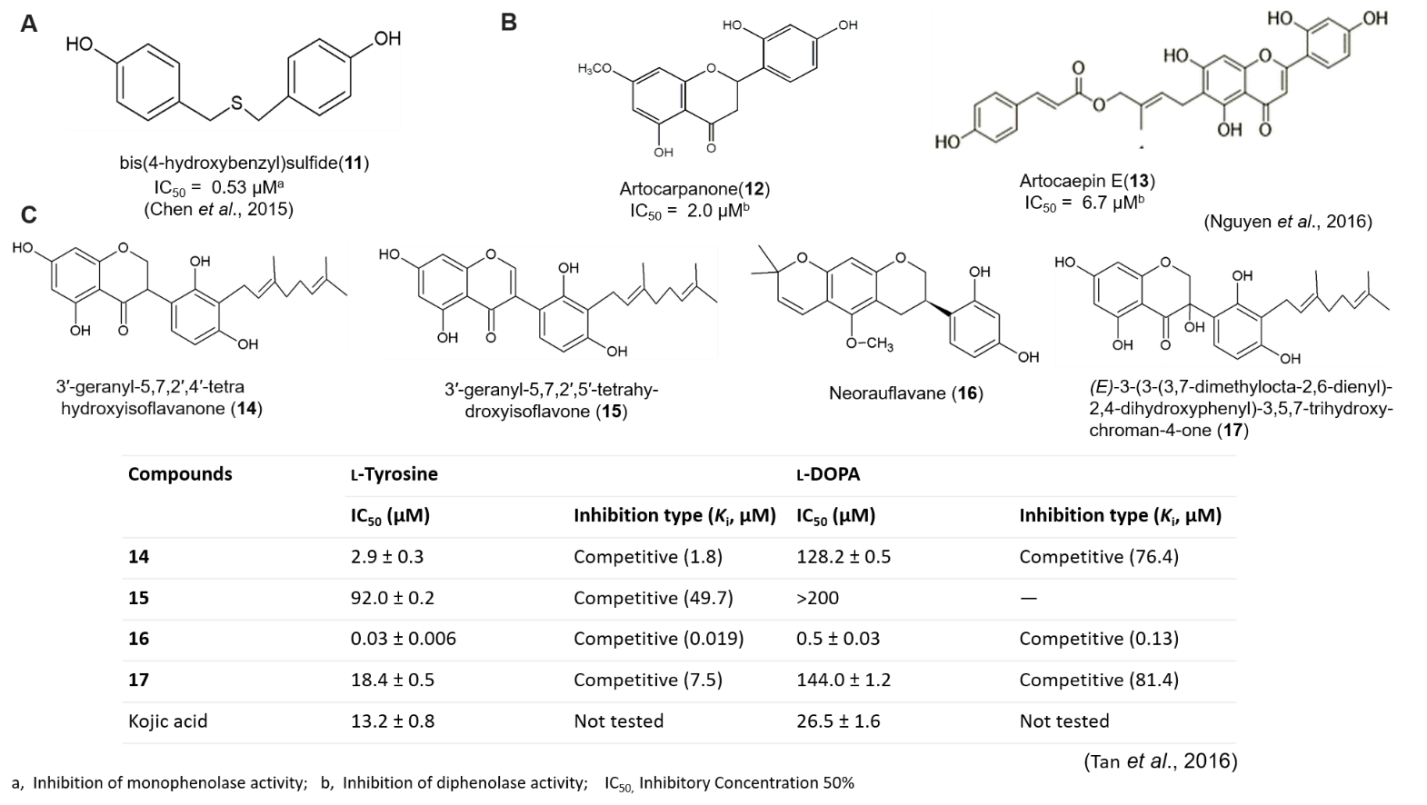
(A) Chemical structure of bis(4-hydroxybenzyl)sulfide isolated from Gastrodia elata
(B) Chemical structure of artocarpanone and artocaepin E isolated from Artocarpus heterophyllous
(C) Chemical structure and tyrosinase inhibitory activity of compounds 14-17 isolated from Campylotropis hirtella
Tan et al. (2016), studied on tyrosinase inhibitory methanol extract of Campylotropis hirtella, and isolated fourteen phenolic compounds, among which neorauflavane emerged as a lead structure for tyrosinase inhibition with IC50 in nanomolar concentration (Figure 9C). Neorauflavane (16) inhibited monophenolase (IC50 = 30 nM), which was 400-fold more active than kojic acid (IC50 = 13.2 μM). The geranylated isoflavanone (14) also inhibited monophenolase and diphenolase activity with IC50values 2.9 and 128.2 μM, respectively. Isoflavanone (17) bearing hydroxyl group on C-3 position inhibited tyrosinase efficiently with 18.4 μM of IC50. Compounds 14 and 16, with best candidate inhibitors among them, manifested competitive, simple reversible slow-binding inhibition against tyrosinase. Moreover, molecular docking analysis showesd that the interaction energies between the enzyme and two inhibitors, compound 14 and 16, were −50.59 kcal/mol and −53.92 kcal/mol, respectively.
Artocarpus heterophyllous (AH), popularly known as jackfruit; and is cultivated for its edible fruits, while the wood has been used for its pharmacological values including anti-tyrosinase activities. Nguyen et al. (2016), reported that flavonoids isolated from MeOH extract of the wood of AH showed potent tyrosinase inhibitory activity. They isolated seven compounds from AH which were artocaepin E, artocaepin F, norartocarpetin, artocarpanone, liquiritigenin, steppogenin and dihydromorin with an IC50 of 6.7 ± 0.8, ˃50, ˃50, 2.0 ± 0.1, 22.0 ± 2.5, 7.5 ± 0.5 and ˃50 μM, respectively . Artocarpanone (Figure 9B) had the most potent tyrosinase inhibitory effect, with an IC50 of 2.0 ± 0.1 μM, followed by artocaepin E, steppogenin and liquiritigenin, with IC50 values of 6.7 ± 0.8, 7.5 ± 0.5 and 22.0 ± 2.5 μM respectively, compare with kojic acid (IC50 44.6 ± 0.4 μM) as positive control. Structure–activity relationship showed that for artocaepin E , the presence of one hydroxyl group at C-2′ and a trans-p-coumaroyl unit connected to the hydroxyprenyl through an ester linkage at C-6 of the flavone skeleton, led to significantly stronger inhibitory activity than that of norartocarpetin. So, the absence of the side-chain at C-6 of the B-ring leads to a significant loss of activity, and the presence of a side-chain such as trans-p-coumaroyl connected to the hydroxyprenyl may positively influence the tyrosinase inhibitory activity. For artocarpanone , which possesses a methoxyl group at C-7 of ring A, had the strongest inhibitory activity. Dihydromorin which has four hydroxyl groups at C-2′, C-3, C-5, and C-7, had weak activity. The study suggested that the methoxyl and hydroxyl groups in the main flavanone skeleton play an important role in tyrosinase inhibition.
Chaita et al.(2017), investigated 900 extracts from Greek plants for potential tyrosinase inhibition properties. Among the five most potent extracts, the methanol extract of Morus alba wood (MAM) demonstrated a significant reduction in intracellular tyrosinase and melanin content in B16F10 melanoma cells. Bioassay-guided isolation led to the acquisition of twelve compounds (Figure 10A). Among these, 2,4,3′-trihydroxydihydrostilbene(24) and dihydrooxyresveratrol(22) constitutes a potent tyrosinase inhibitor with an IC50 0.8 ± 0.15 and IC50 0.3 ± 0.05 compare to kojic acid(IC50 16.1 ± 1.4). MAM extract and compounds oxyresveratrol(18), trans-dihydromorin(23) and 2,4,3′-trihydroxydihydrostilbene(24) also significantly suppress in vivo melanogenesis during zebrafish embryogenesis. Two dihydrostilbenes, dihydrooxyresveratrol(22) and 2,4,3′-trihydroxydihydrostilbene(24) showed the stronger inhibition than their stilbene analog, oxyresveratrol (IC50 1.7 μM). Study suggested that the enhancement of the tyrosinase inhibitory properties may be responsible for the reduction of the double bond in the stilbene structure, as the subsequent bibenzyl structure allows the molecule to approach and interact with the active center more effectively.

(A) Compounds isolated from Morus alba and their tyrosinase inhibitory activity
(B) Chemical structure of oxyresveratrol, dihydrooxyresveratrol, trans-dihydromorin and 2,4,3′-trihydroxydihydrostilbene isolated from Morus alba
Condensed tannins are important class of plant-derived natural products and have potential to be leads for new drugs due to their biological activities such as antioxidant, anticancer, antimicrobial and anti-cardiovascular activities. Condensed tannins are composed of flavan-3-ol sub-units linked mainly through C4→C8 (or C4→C6) bonds. Chen et al. (2014), isolated condensed tannins from Ficus virens (F. virens) leaves, fruit, and stem bark. The extracts were prepared using acetone as a solvent. The condensed tannins from different extracts of F. virens, were complex mixtures of homo- and heteropolymers of B-type procyanidins and prodelphinidins(11A) with degrees of polymerization up to hexamer, dodecamer, and pentadecamer, respectively. The condensed tannins exhibited efficiency in inhibiting both mono- and diphenolase activities of the mushroom tyrosinase, and were found to be mixed type inhibitors of the enzyme. The IC50 value for the leaves, fruit, and stem bark condensed tannins determined to 131.67, 99.89, and 106.22 μg/ml on monophenolase activity, and 128.42, 43.07, and 74.27 μg/ml on diphenolase activity (Figure 11B). Moreover, fluorescence quenching, copper interacting, and molecular docking techniques were utilized to find out the molecular mechanisms of the inhibition and the results showed that the inhibition was carried out mainly through the interaction of the hydroxyl groups in the aromatic ring B of the condensed tannins with the active center of the enzyme by chelating dicopper ions. Additionally, the condensed tannins could directly scavenge the o-quinones through oxidoreduction reaction. This study group suggested that, the condensed tannins could be used to design and screen for potent novel tyrosinase inhibitors. Recently, another study (Chai et al., 2018) suggested that condensed tannins from longan bark might be a good source of tyrosinase inhibitor and could be used as novel food preservatives and medicines of skin diseases. Results showed good inhibitions on proliferation, intracellular enzyme activity and melanogenesis of mouse melanoma cells.

(A) Chemical structure of the condensed tannins (B) Inhibition constants of condensed tannins from the leaves, fruit, and stem bark of F. virens. (Chen et al., 2017)
Recently, Wang et al. (2018) studied on the inhibitory effect and mechanism of lignin on tyrosinase activity and founded lignin as a novel tyrosinase inhibitor. In nature, lignin is the most abundant aromatic biopolymer. This study isolated six lignin samples by alkali and ethanol organosolv processes from three typical lignocellulosic feedstocks. The ethanol organosolv lignins showed stronger anti-tyrosinase activity than alkali lignins. For lignins from different sources, corn stalk lignin presented highest inhibitory effect with an IC50 value of 0.276 mg/mL, compare to that of positive control p-hydroxy benzaldehyde (0.233 mg/mL). Moreover, the kinetics study showed that the ethanol organosolv lignin from corn stalk was a reversible mixed-type inhibitor. This study group suggested that lignin possesses antityrosinase activity and can be potentially used as an enzyme inhibitor in overtyrosinase activity control fields.
Tyrosinase inhibition is a well-established strategy for controlling melanin production in vivo and recently the development of human tyrosinase inhibitors raises considerable interest in dermocosmetics. There is significant differences at cellular and structural levels among the tyrosinases from distinct sources, especially plant, mushroom, bacterial, and human (Fogal et al., 2015). Mushroom tyrosinase and bacterial tyrosinase are generally soluble oligomeric enzymes present in the cytosol, while human tyrosinase exists as a highly glycosylated monomeric melanosomal transmembrane protein (van Gelder et al., 1997; Wang and Hebert, 2006). There is a significant differences between mushroom and human tyrosinase enzyme in respect to the catalytic activities and substrate specificities (Hearing et al., 1980). An abundant literature is dedicated to mushroom tyrosinase inhibitors for human applications but unfortunately, currently used inhibitors lack the affinity and selectivity required for human tyrosinase targeting applications. Moreover, harmful toxicity has often been reported (Haudecoeur et al., 2016). Therefore, there is an urgent need for novel selective human tyrosinase inhibitors that match efficacy and safety standards required for the development of products aimed to human use.
Recently, researchers are trying to identify tyrosinase inhibitors targeting human tyrosinase (hTYR). Pillaiyar et al.(2017b) reviewed on medicinal perspective of tyrosinase inhibitors, and discussed about the inhibitory potency and mechanisms of thujaplicins (α, β and γ isomers), 4-butyl resorcinol, linderanolide B and subamolide A, as a human tyrosinase inhibitor. To find out novel human tyrosinase inhibitor, Yoshimori et al., (2014) studied the inhibitory effects of three isomers of thujaplicin (α, β, and γ) on human tyrosinase and analyzed their binding modes using homology model and docking studies, and found that γ-thujaplicin (IC50 = 1.15 μM) is the most potent inhibitor of human tyrosinase among thujaplicins compare to a well-known tyrosinase inhibitor kojic acid (IC50 = 571.17 μM). Comparison of inhibitory activities of thujaplicins and kojic acid against hTYR and mTYR is illustrated in Figure 12A.
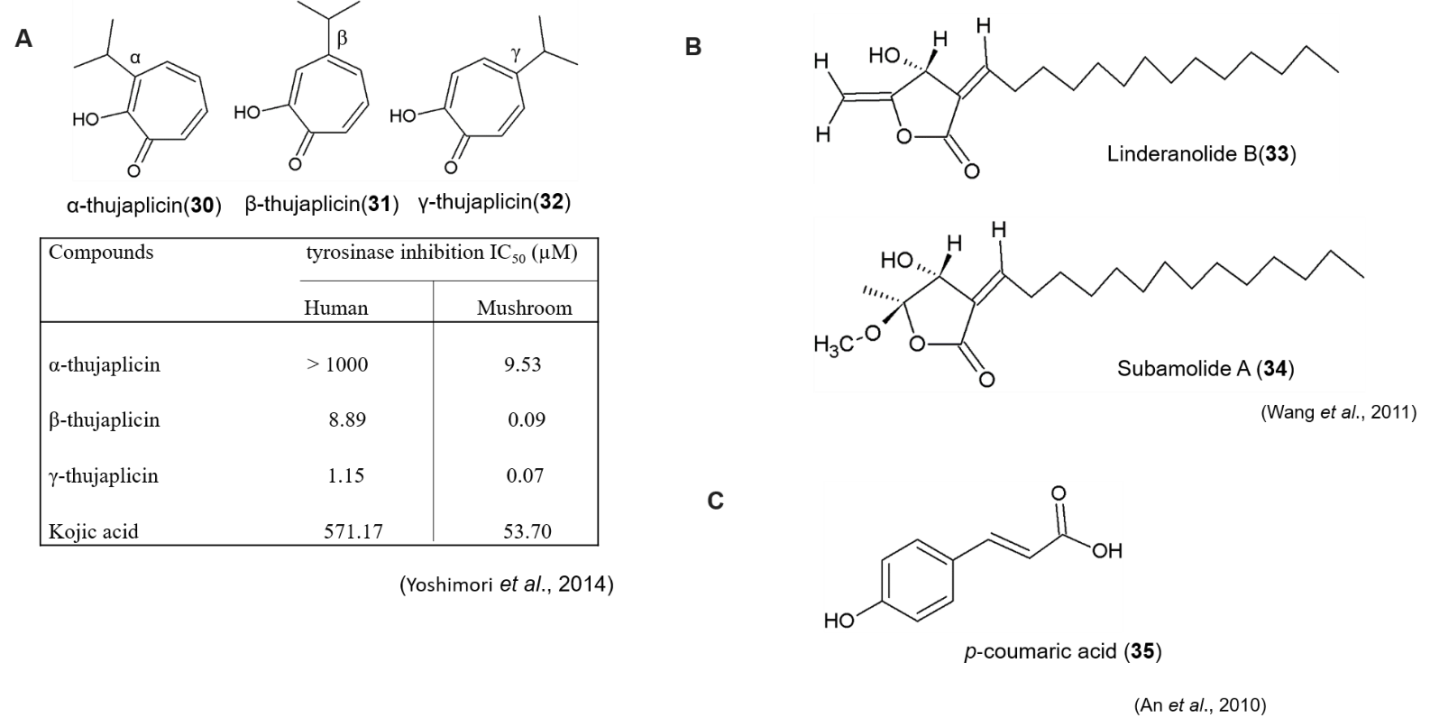
(A) Chemical structure and tyrosinase inhibitory activity of thujaplicin analagoues
(B) Chemical structure of Linderanolide B and Subamolide A
(C) Chemical structure of p‐coumaric acid
Wang et al.,(2011) isolated linderanolide B and subamolide A from the stems of Cinnamomum subavenium. Both of the compounds exhibited mushroom tyrosinase inhibition and also non-cytotoxic to normal human skin cells and zebrafish system. Linderanolide B and subamolide A (Figure 12B) reduced human tyrosinase activities (50%) at a dose of 1μm after 48h of treatment and effectively inhibited the melanin production in HEMn‐MP cells (40% reduction). Through molecular docking, in a virtual model of human tyrosinase, it revealed that both of the compounds block copper ions within the tyrosinase active site. This study suggested that linderanolide B and subamolide A are effective novel tyrosinase inhibitors to be considered as skin‐lightening agents.
In another study, An et al.(2010) compared the inhibitory effects of p‐coumaric acid (p‐CA), arbutin and kojic acid, on the catalytic activities of mushroom, murine and human tyrosinases in vitro, using tyrosine and 3,4‐dihydroxyphenylalanine as substrates. The results showed that p‐CA (Figure 12C) is a stronger inhibitor of human or murine than mushroom tyrosinase, in comparison with kojic acid and arbutin as a positive control. Moreover, p‐CA inhibited human TYR at much lower concentrations than those required for the inhibition of murine or mushroom TYRs.
4-Butyl resorcinol, a resorcinol derivative that has an inhibitory effect on TYR and TRP-1 , was introduced in 1995 as a hypopigmenting agent (Okubo et al., 1995). Kolbe et al.(2013) examined the inhibitory effectsof4-butyl resorcinolonhTYRcompare with kojic acid, hydroquinone and arbutin as positive control. 4‐butylresorcinol with an IC50 of 21 μmol/L represented one of the potent inhibitor of human tyrosinase. 4-Butyl resorcinol exhibited 20-times more-potent inhibitory activity than kojic acid, which showed an IC50 of 500 μM. Arbutin and hydroquinone only weakly inhibit human tyrosinase with a IC50 in the millimolar range.
Aurones (2-benzylidenebenzofuran-3(2H)-ones), naturally occurring flavonoids act as inhibitors of melanin biosynthesis in human melanocytes. It also act as effectors of mushroom tyrosinase and as molecular probes for the investigation of the binding-site structural homology between mushroom and bacterial tyrosinase (Haudecoeur et al., 2016). On the other hand, HOPNO, a catechol-mimicking, nonoxidizable moiety, is a potent inhibitor of mushroom tyosinase (Ki = 1.8 μM). Haudecoeur et al.(2016) synthesized aurone derivatives (36-38) whose B-ring replaced by a non-oxidizable 2-hydroxypyridine-N-oxide (HOPNO) moiety (Figure 13). The synthesized compounds tested for tyrosinase and melanogenesis inhibitory activity using purified human tyrosinase and human melanoma MNT-1 cells, respectively, compare with kojic acid as positive control. The result showed that compounds 37 and 38 shared a similar inhibition potency (Ki = 1.02 and 1.2 μM, respectively), whereas analogue 36 (Ki = 0.35 μM) was found 3.5 times more active (Figure13). Compound 36 (IC50 = 16.6 μM) showed better efficiency in suppressing melanogenesis in MNT-1 cells compare to others. This study suggested that HOPNO-embedded 6-hydroxyaurone is one of the best effective inhibitor of isolated human tyosinase.

Chemical structure and tyrosinase inhibitory activity of aurones derivatives
Mann et al. (2018), used recombinant human tyrosinase to screen a library of 50,000 compounds and compared the active screening hits with well-known whitening ingredients. Among them, resorcinol derivatives thiamidol (isobutylamido thiazolyl resorcinol), 4-butylresorcinol, 4-hexylresorcinol, and 4-phenylethylresorcinol were the most promising inhibitors of the diphenolase (l-dopa oxidase) activity of human tyrosinase with IC50 values of 1.1 μmol/L, 21 μmol/L, 94 μmol/L, and 131 μmol/L, respectively (Figure 14). Kojic acid, a well-known tyrosinase inhibotor was 500 times less potent than thiamidol, with an IC50 of about 500 μmol/L. But, thiamidol only weakly inhibited mushroom tyrosinase with an IC50 value of 108 μmol/L. kinetic analysis of the inhibition of human tyrosinase by thiamidol yielded a strictly competitive type of inhibition with an inhibitor constant (Ki) of 0.25 μmol/L, whereas the Ki values for 4-butylresorcinol (9 μmol/L), 4-hexylresorcinol (39 μmol/L), and 4-phenylethylresorcinol (24 μmol/L) were markedly higher than the Ki value of thiamidol. Thiamidol strongly and reversibly inhibited melanin production (IC50 = 0.9 μmol/L) in MelanoDerm skin models. In monolayer cultures, it visibly reduced melanin formation. 4-Butylresorcinol and hydroquinone inhibited melanin synthesis in MelanoDerm skin models with an IC50 of 13.5 and 15 μmol/L, respectively. In long-term melanocyte monolayer cultures, thiamidol (1μmol/L) also reduced melanin production to less than 60% after 2 weeks. Virtual docking studies revealed the possible binding modes of thiamidol to human tyrosinase, and found a strong hydrophobic subpocket mainly by the side chains of I368, V377, and F347. The 1-hydroxy group of the aromatic ring of thiamidol makes extensive contacts with the di-copper center of human tyrosinase, and the 3-hydroxy group is involved in hydrogen bonds with the side chain of S380. To examine the in vivo efficacy of thiamidol, a clinical study was also carried out. The elderly subjects treated age spots on their skin twice daily with a formula containing 0.2% thiamidol or with the vehicle only as a control. After 4 weeks of treatment, the treated age spots were significantly lighter than the untreated control age spots and more improvement continued over the 12 weeks of treatment. A follow-up study showed that concentrations of thiamidol as low as 0.1% effectively reduced the visibility of age spots. From this study it was found that thiamidol (isobutylamido thiazolyl resorcinol) was a potent inhibitor of human tyrosinase with a remarkable efficacy in vitro and in vivo. But the efficacy is distinctively different in mushroom tyrosinase, where 4-butylresorcinol, 4-hexylresorcinol, and 4-phenylethylresorcinol, and even kojic acid, are superior to thiamidol in inhibiting the enzyme (Figure 14). The 4-substituted resorcinol motif has been known for an efficient chemical moiety that inhibits tyrosinase activity. From structure-activity relationship, the thiazolylamide moiety of thiamidol conveys a much better inhibition of human tyrosinase than do the hydrocarbon side chains present in three other derivatives of resorcinol (4-butyl-, 4-hexyl-, and 4-phenyl ethylresorcinol).

Chemical structure and tyrosinase inhibitory activity of thiamidol and resorcinol derivatives
The impact of cosmetic skin concerns on quality of life and the abnormal pigmentation causes serious esthetic problems as well as life threating diseases. Hyperpigmentation is an important issue and the most prominent target for inhibiting hyperpigmentation is tyrosinase, the rate-limiting enzyme in melanogenesis. Tyrosinase inhibitors have a huge demand in cosmetic and medicinal industries due to their preventive effect on pigmentation disorders as well as skin-whitening effect. A number of tyrosinase inhibitors have been identified from both natural and synthetic sources, but only a few of them are used as skin-whitening agents, primarily due to various safety concerns such as cytotoxicity, solubility, effective cutaneous absorption, etc. Many tyrosinase inhibitors presently used as skin-whitening agents have found specific drawbacks including carcinogenicity. According to the WHO, skin lighteners are found to cause cancers in human, as well as some manufacturers employ harmful toxins such as mercury in skin lightening products to block melanin production (www.globenewswire.com). So, consumers are now inclining towards adoption of organic and natural products. Moreover, there is a significant differences between mushroom and human tyrosinase enzyme in respect to the catalytic activities and substrate specificities. Currently used inhibitors that was identified using mushroom tyrosinase, lack the affinity and selectivity required for human tyrosinase. Therefore, there is an urgent need for novel selective human tyrosinase inhibitors that match efficacy and safety standards required for the development of products aimed to human use including validation of skin-whitening efficacy. So, more concrete studies with a human clinical point of view are required for the found inhibitors.