Abstract
Mutations of isocitrate dehydrogenase 1 (IDH1) and isocitrate dehydrogenase 2 (IDH2) have been reported in gliomas, cartilaginous tumors, and acute myeloid leukemias. IDH mutations are specific to a single codon in the conserved and functionally important arginine 132 residue (R132) of IDH1 or arginine 172 residue (R172) of IDH2 in gliomas. Although IDH1 and IDH2 catalyze the oxidative carboxylation of isocitrate to α-ketoglutarate in cytosol and mitochondria, respectively, mutated IDH1/2 proteins can possess the ability to change α-ketoglutarate to an oncometabolite R(−)-2-hydroxyglutarate. We have established several monoclonal antibodies (mAbs) specific for IDH1/2 mutations. However, no multi-specific mAb against IDH1/2 mutations has been reported. For this study, we immunized mice with an IDH1-R132G peptide of 19 amino acids (GGVKPIIIGGHAYGDQYRA), and established a novel mAb MsMab-1 that recognizes IDH1-R132G, but not wild type IDH1 in enzyme-linked immunosorbent assay (ELISA). It is particularly interesting that MsMab-1 recognizes all IDH1 mutants (R132H, R132C, R132S, R132G, R132L) in ELISA. Western blot analysis also revealed that MsMab-1 reacted with recombinant proteins of IDH1-R132H, IDH1-R132S, and IDH1-R132G, but not with wild type IDH1 and other IDH1 mutations, indicating that MsMab-1 is a multi-specific anti-mutated IDH1 mAb. Unexpectedly, MsMab-1 recognizes IDH2-R172M protein, despite that the IDH1-R132G peptide shows only 73.7% identity with the equivalent portion of IDH2-R172M (GGTKPITIGMHAHGDQYKA). Moreover, MsMab-1 stained the IDH1-R132S or IDH1-R132G-expressing glioma cells in immunohistochemistry. This report is the first to establish a multi-specific anti-mutated IDH1/2 mAb, that is expected to be useful for immunohistochemical determination of IDH1/2 mutation-bearing tumors.
Introduction
Mutations of isocitrate dehydrogenase 1 (IDH1) and isocitrate dehydrogenase 2 (IDH2) were found to create the capability of the enzyme to change α-ketoglutarate to an oncometabolite, R(−)-2-hydroxyglutarate (2-HG), although IDH1 and IDH2 catalyze the oxidative carboxylation of isocitrate to α-ketoglutarate in cytosol and mitochondria, respectively (Dang et al. 2009). Reportedly, 2-HG accumulates in an inherited metabolic disorder, 2-hydroxyglutaric aciduria, as a consequence of 2-hydroxyglutarate dehydrogenase deficiency, because 2-hydroxyglutarate dehydrogenase converts 2-HG to α-ketoglutarate (Struys et al. 2005). Patients with 2-hydroxyglutarate dehydrogenase deficiencies are known to accumulate 2-HG in the brain, to develop leukoencephalopathy, and to have increased risk of brain tumors. Moreover, elevated 2-HG levels in the brain result in increased reactive oxygen species (ROS) concentrations, potentially contributing to an increased risk of cancer (Kolker et al. 2002). IDH1/2 mutations occur in some gliomas (Yan et al. 2009), cartilaginous tumors (Amary et al. 2011; Pansuriya et al. 2011), and in acute myeloid leukemias (Ward et al. 2010). In astrocytomas, oligodendrogliomas, oligoastrocytomas, and secondary glioblastomas, IDH1/2 mutations have been identified as early and frequent genetic alterations (50-93%). They might be the initiating event in these glioma subtypes (Balss et al. 2008; Parsons et al. 2008; Sonoda et al. 2009; Yan et al. 2009). In contrast, primary glioblastomas rarely contain IDH1/2 mutations (< 5%).
The IDH1 mutations are remarkably specific to a single codon in the conserved and functionally important arginine 132 residue (R132). In contrast, the IDH2 mutations are specific to a single codon in arginine 172 residue (R172) in gliomas. It is particularly interesting that IDH2 mutations of AML were discovered subsequently in arginine 140 residue (R140), which is more frequent than R172 (Paschka et al. 2010). The vast majority of changes are heterozygous. In gliomas, IDH1 mutations were reported as IDH1-R132H (664/716: 92.7%), IDH1-R132C (29/716: 4.2%), IDH1-R132S (11/716: 1.5%), IDH1-R132G (10/716: 1.4%), and IDH1-R132L (2/716: 0.2%) (Hartmann et al. 2009). Although one monoclonal antibody (mAb) against IDH1-R132H has been reported (Capper et al. 2009), we established IDH1-R132H-specific mAbs IMab-1/HMab-1 (Kato et al. 2009; Takano et al. 2012), IDH1-R132S-specific mAb SMab-1 (Kaneko et al. 2011), and IDH1-R132G-specific mAbs GMab-r1/GMab-m1 (Kaneko et al. 2013a; Kato et al. 2013). In contrast, IDH2 mutations were reported as IDH2-R172K (20/31: 64.5%), IDH2-R172M (6/31: 19.3%), and IDH2-R172W (5/31: 16.2%) in gliomas (Hartmann et al. 2009). To date, we have established IDH2-R172K-specific mAb KMab-1 (Kaneko et al. 2013b), IDH2-R172M-specific mAb MMab-1 (Kaneko et al. 2013b), and IDH2-R172W-sepcific WMab-1 (Kato and Kaneko 2013). In the present report, we describe a novel multi-specific anti-mutated IDH1/2 monoclonal antibody, MsMab-1, that is expected to be extremely useful for diagnosis of IDH1/2 mutation-bearing tumors.
Materials and Methods
Cell lines and tissues
Chinese hamster ovary (CHO)-K1 cells (CCL-61) and Sp2/0-Ag14 myeloma (CRL-1581) and were obtained from the American Type Culture Collection (ATCC, Manassas, VA). These cell lines were cultured at 37°C in a humidified atmosphere of 5% CO2 and 95% air in RPMI 1640 medium including 2 mM L-glutamine (Nacalai Tesque, Inc., Kyoto, Japan) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Life Technologies Corp., Carlsbad, CA). This study included one glioma (oligodendroglioma, WHO grade II) who underwent primary surgery at Nagoya University Hospital (Kato et al. 2013) and two glioma patients (glioblastoma, WHO grade IV) who underwent primary surgery at Tsukuba University Hospital (Takano et al. 2012). Ethical committee of each university approved our study. Informed consent for obtaining samples and for subsequent data analysis was obtained from each patient or the patient’s guardian.
Hybridoma Production
MsMab-1 was produced using mouse medial iliac lymph node methods (Kaneko et al. 2013b) Institutional animal committee approved our study. Briefly, BDF1 mice (Japan SLC Inc., Shizuoka, Japan) were immunized by injecting 80 μg of synthetic peptides of CGGVKPIIIGGHAYGDQYRA (IDH1-R132G), conjugated with KLH together with Freund’s complete adjuvant (FCA; Sigma-Aldrich Corp., St. Louis, MO) into the tailbase. The lymphocytes were fused with mouse myeloma Sp2/0-Ag14 cells using polyethylene glycol (PEG). The culture supernatants were screened using ELISA for binding to the IDH1-R132G and IDH1-WT peptides, conjugated with BSA.
Enzyme-Linked Immunosorbent Assay (ELISA)
Synthetic peptides were immobilized, respectively, on Nunc Maxisorp 96-well immunoplates (Thermo Fisher Scientific Inc., Waltham, MA) at 1 μg/ml for 30 min. Synthetic peptides (Sigma-Aldrich Corp.) are as follows: GGVKPIIIGRHAYGDQYRA (IDH1-WT), GGVKPIIIGHHAYGDQYRA (IDH1-R132H), GGVKPIIIGCHAYGDQYRA (IDH1-R132C), GGVKPIIIGSHAYGDQYRA (IDH1-R132S), GGVKPIIIGGHAYGDQYRA (IDH1-R132G), GGVKPIIIGLHAYGDQYRA (IDH1-R132L). After blocking with SuperBlock T20 (PBS) Blocking Buffer (Thermo Fisher Scientific Inc.), the plates were incubated with culture supernatant or purified mAbs (1 μg/ml) with subsequent 1:1,000 diluted peroxidase-conjugated anti-mouse IgG (Dako, Glostrup, Denmark). The enzymatic reaction was conducted with 1-Step Ultra TMB-ELISA (Thermo Fisher Scientific Inc.). The optical density was measured at 655 nm using an iMark microplate reader (Bio-Rad Laboratories Inc., Philadelphia, PA). These reactions were performed with a volume of 50 μl at 37°C.
Plasmid preparation
Human IDH1 cDNA (GenBank accession no. AF113917 or BC012846) or human IDH2 cDNA (accession no. NM_002168) encoding a full length open reading frame (ORF) was obtained by PCR using a human lung cDNA library (Cosmo Bio Co. Ltd., Tokyo, Japan) or cDNA derived from the U373 glioblastoma cell line as template, respectively. The primer set for IDH1 was as follows: EcoRI-IDH1-F1, 5′-cacgaattcATGTCCAAAAAAATCAGTGG-3′ and SalI-IDH1-R1, 5′-gtggtcgacTTAAAGTTTGGCCTGAGCTA-3′. The primer set for IDH2 was as follows: EcoRI-IDH2.F1, 5′-ccgaattcgggATGGCCGGCTACCTGCGGG-3′ and SalI-IDH2wterR1359, 5′-gccgtcgacCTACTGCCTGCCCAGGGCTCT-3′. The amplified cDNA was subcloned into a pcDNA3.1/V5-His-TOPO vector (Life Technologies Inc.). Substitution of the arginine 132 (R132) to appropriate amino acid codons in IDH1 or arginine 172 (R172) to appropriate amino acid codons in IDH2 was introduced using the QuikChange Lightning site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA). The full-length IDH1/2 were subcloned into an expression vector, pMAL-c2 (New England Biolabs Inc., Beverly, MA) via EcoRI and SalI restriction sites. The full-length IDH2 and each mutated ORF were amplified using the primer set: EcoRI-IDH2.F1 and IDH2woterR1356-XhoI, 5′- taactcgagcgCTGCCTGCCCAGGGCTCTG-3′. These PCR products were digested with EcoRI and XhoI restriction enzymes, and were subcloned into pcDNA3 vector (Life Technologies Inc.) together with the nucleotide sequence (ctcgagTGGCGTTGCCATGCCAGGTGCCGAAGATGATGTGGTGTAAtctaga), which encodes 12 amino acids, GVAMPGAEDDVV (PA tag) (Kaneko et al. 2013b; Kato and Kaneko 2013).
Protein expression using bacteria cells and mammalian cells
Competent Escherichia coli TOP-10 cells (Life Technologies Inc.) were transformed with a plasmid, pMAL-IDH1-WT, pMAL-IDH1-R132H, pMAL-IDH1-R132C, pMAL-IDH1-R132S, pMAL-IDH1-R132G, pMAL-IDH1-R132L, pMAL-IDH2-R172K, pMAL-IDH2-R172M, or pMAL-IDH2-R172W. Then, they were cultured overnight at 37°C in LB medium (Life Technologies Inc.) containing 100 μg/ml ampicillin (Sigma-Aldrich Corp.). Cell pellets were resuspended in phosphate buffered saline (PBS) with 1% Triton X-100 with 50 μg/ml aprotinin (Sigma-Aldrich Corp.). After sonication using Branson Advanced Sonifier (Branson Ultrasonics Corp., Danbury, CT), the crude extracts were collected by centrifugation (9,000 × g, 30 min, 4°C). The supernatants were loaded onto amylose resin (New England Biolabs Inc.). The loaded resins were washed extensively with column buffer (20 mM Tris-HCl, pH 7.4, 200 mM NaCl, 1 mM EDTA). The fusion proteins were eluted with column buffer containing 10 mM maltose. Then CHO cells were transfected with an appropriate amount of each plasmid, pcDNA3.1/IDH1-WT, pcDNA3.1/IDH1-R132H, pcDNA3.1/IDH1-R132C, pcDNA3.1/IDH1-R132S, pcDNA3.1/IDH1-R132G, pcDNA3.1/IDH1-R132L, pcDNA3/IDH2-R172K, pcDNA3/IDH2-R172M, or pcDNA3/IDH2-R172W using Lipofectamine LTX (Life Technologies Inc.) according to the manufacturer’s instructions. The expression level of IDH1/2 was confirmed using Western blot analysis.
Western blot analyses
Cultured cell pellets were lysed with PBS with 1% TritonX-100 for 30 min on ice. The lysate supernatants were centrifuged for 15 min at 15,000 rpm to remove cellular debris. Cell lysates containing 5 μg of total protein or 0.05 μg of purified recombinant proteins were prepared for Western blot analyses by boiling in SDS sample buffer (Nacalai Tesque, Inc.). They were electrophoresed on 5-20% polyacrylamide gels (Wako Pure Chemical Industries Ltd.). The separated proteins were transferred to a PVDF membrane (EMD Millipore Corp., Billerica, MA). After blocking with 4% skim milk in PBS with 0.05% Tween20 for 15 min, the membrane was incubated with 1 μg/ml of MsMab-1, HMab-1 (Takano et al. 2012), SMab-1 (Kaneko et al. 2011), GMab-r1 (Kaneko et al. 2013a), KMab-1 (Kaneko et al. 2013b), MMab-1 (Kaneko et al. 2013b), WMab-1 (Kato and Kaneko 2013), RcMab-1 (Kaneko et al. 2013a; Kato et al. 2013), KrMab-3 (Kaneko et al. 2013b), anti-MBP tag (TMab-2) (Kaneko et al. 2013a, b; Kato et al. 2013; Kato and Kaneko 2013), anti-PA tag (NZ-1) (Kaneko et al. 2013b; Kato and Kaneko 2013), and anti-V5 tag (MBL Co. Ltd., Nagoya, Japan) for 30 min. Then the membrane was incubated with peroxidase-conjugated secondary antibodies (1:1,000 diluted; Dako) for 15 min, and developed with Pierce Western Blotting Substrate Plus (Thermo Fisher Scientific Inc.) using Sayaca-Imager (DRC Co. Ltd., Tokyo, Japan).
Immunohistochemical analysis
Mutated IDH1/2 protein expression was determined immunohistochemically in paraffin-embedded tumor specimens. Briefly, 4-μm-thick histologic sections were deparaffinized in xylene and rehydrated. Then, they were autoclaved in citrate buffer (pH 6.0; Dako) for 20 min. Sections were incubated with 5 μg/ml of MsMab-1 overnight at 4°C. Biotin-conjugated secondary anti-mouse IgG (Dako) was incubated for 30 min at room temperature followed by the peroxidase-conjugated biotin-streptavidin complex (Vectastain ABC Kit; Vector Laboratories Inc., Burlingame, CA) for 30 min at room temperature. Color was developed using 3, 3-diaminobenzidine tetrahydrochloride (DAB; Dako) for 10 min, and counterstained with hematoxylin.
Results
Production of mutated IDH1/2-specific monoclonal antibodies
Multi-specific antibodies against mutated IDH1/2 have not been reported, although we previously established several anti-mutated IDH1/2 mAbs. As shown in Table 1, anti-IDH1 mAbs (HMab-1, SMab-1, GMab-r1) recognized each mutation-containing peptide, indicating that HMab-1, SMab-1, and GMab-r1 are mono-specific. For this study, mice were immunized with synthetic peptides of IDH1-R132G, and the wells of hybridomas, which produced IDH1-R132G-reactive / IDH1-wild type (WT)-nonreactive antibodies, were screened in ELISA. After limiting dilution, clone MsMab-1 (mouse IgG2a, kappa) against IDH1-R132G was established. It is noteworthy that MsMab-1 was shown to be reactive with all peptides of IDH1 mutation (IDH1-R132H, IDH1-R132C, IDH1-R132S, IDH1-R132G, and IDH1-R132L) in ELISA, but not with IDH-WT peptide, whereas RMab-3 recognized all IDH1 peptides (Table 1).
Specificity of MsMab-1 against IDH1/2 mutants
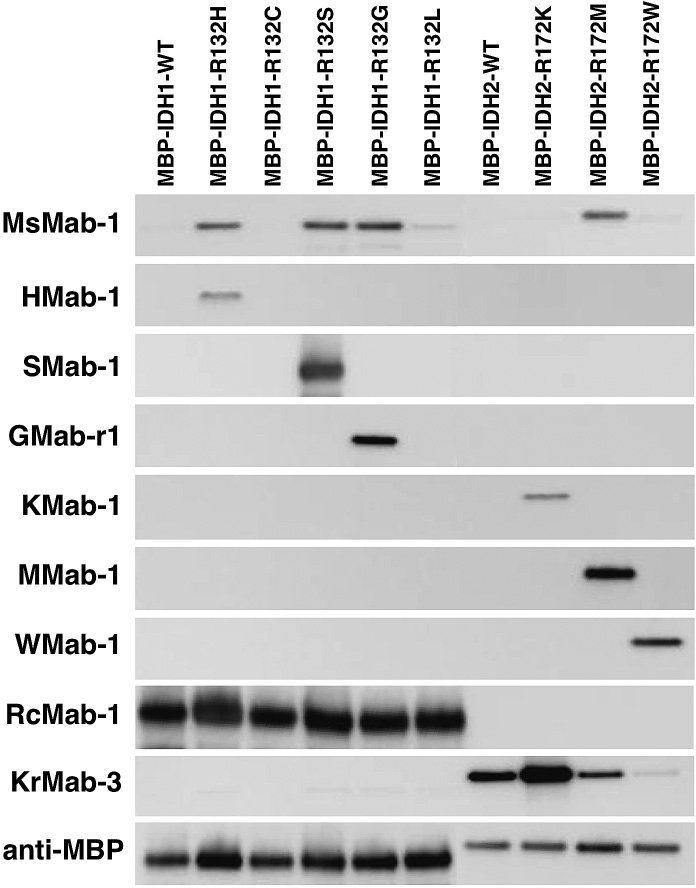
To determine the specificity of MsMab-1 further, we investigated the reactivity against IDH1-WT and IDH1 mutants (MBP-IDH1-R132H, MBP-IDH1-R132C, MBP-IDH1-R132S, MBP-IDH1-R132G, MBP-IDH1-R132L) using Western blot analyses. Fig. 1 shows that MsMab-1 strongly recognized MBP-IDH1-R132H, MBP-IDH1-R132S, and MBP-IDH1-R132G proteins, but only weakly reacted with MBP-IDH1-R132L, indicating that MsMab-1 is a multi-specific antibody against mutated IDH1 proteins. In contrast, HMab-1, SMab-1, GMab-r1, KMab-1, MMab-1, and WMab-1 recognized each mutation. RcMab-1 recognized MBP-IDH1-WT and all mutated MBP-IDH1 proteins. It is particularly interesting that MsMab-1 also reacted with MBP-IDH2-R172M, but not with MBP-IDH2-WT, although the IDH1-R132G peptide of 19 amino acids, which was used for immunization, shows only 73.7% identity with the corresponding amino acid sequence of the IDH2-R172M peptide (GGTKPITIGMHAHGDQYKA). KrMab-3 showed the strong reaction with MBP-IDH2-WT, MBP-IDH2-R172K, and MBP-IDH2-R172M, and weak reaction with MBP-IDH2-R172W. These results indicate that MsMab-1 is useful for detecting IDH1-R132H/R132S/R132G and IDH2-R172M proteins.
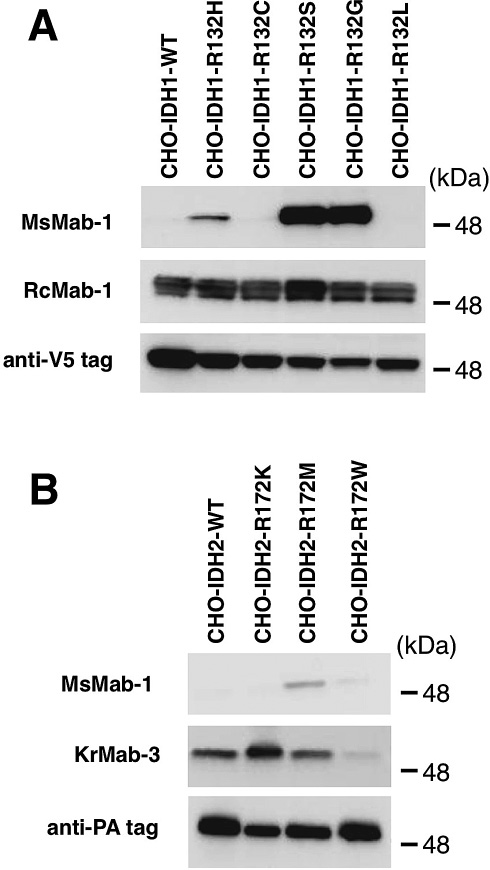
We next performed Western blot analyses against mutated IDH1/2-expressing CHO cells. As presented in Fig. 2A, MsMab-1 strongly recognized IDH1-R132S and IDH1-R132G proteins expressed in CHO cells; weakly reacted with IDH1-R132H. It did not react with the other proteins (IDH1-WT, IDH1-R132C, IDH1-R132L), which indicated that MsMab-1 is useful for detecting IDH1-R132H/R132S/R132G expressed in mammalian cells. In contrast, RcMab-1 equally recognized IDH1-WT and all IDH1 mutations (upper band). RcMab-1 also detected the endogenous hamster IDH1 of CHO cells (lower band). As presented in Fig. 2B, MsMab-1 reacted with IDH2-R172M expressed in CHO cells, not with IDH2-WT or other IDH2 mutations. KrMab-3 showed strong reaction with IDH2-WT, IDH2-R172K, and IDH2-R172M, but only weak reaction with IDH2-R172W.
Immunohistochemical analysis by MsMab-1 against IDH1-R132S or IDH1-R132G-bearing gliomas
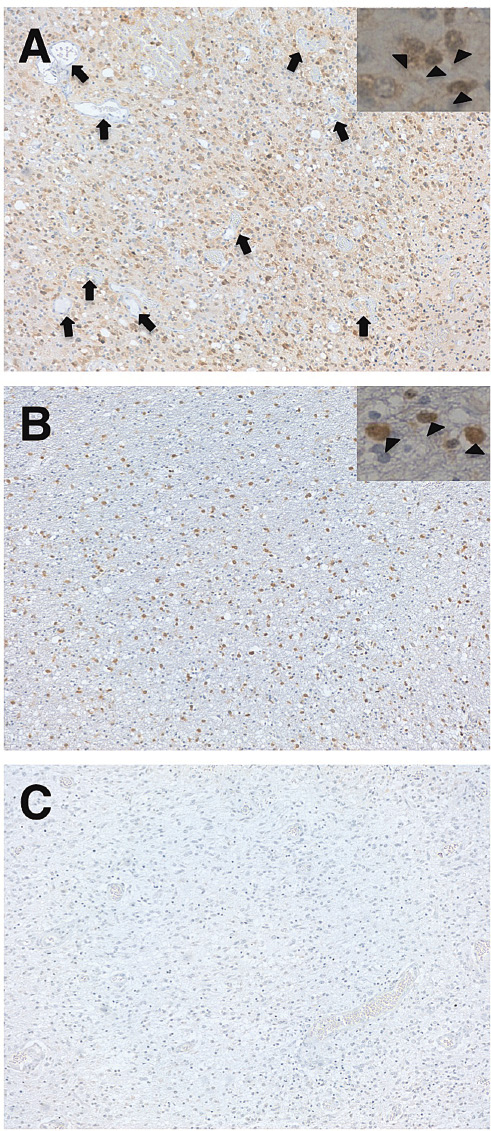
We next performed immunohistochemistry of MsMab-1 against IDH1-R132S-positive or IDH1-R132G-positive gliomas (Fig. 3) because MsMab-1 strongly recognized IDH1-R132S and IDH1-R132G proteins expressed in mammalian cells (Fig. 2A). MsMab-1 stained almost all tumor cells of IDH1-R132S-positive gliomas (Fig. 3A) and IDH1-R132G-positive gliomas (Fig. 3B), although no staining was observed in IDH1/2-WT gliomas (Fig. 3C). MsMab-1 stained not only cytoplasm but also nucleus of glioblastoma cells (inset of Fig. 3A) or oligodendroglioma cells (inset of Fig. 3B). This cytoplasmic/nuclear staining pattern of MsMab-1 is coincident with that described in previous reports, which demonstrates that an anti-IDH1-R132H mAb stains both cytoplasm and nucleus of glioma cells, although wild type IDH1 is expressed in cytosol (Ikota et al. 2011). Moreover, MsMab-1 did not stain endothelial cells in gliomas, indicating that MsMab-1 only stained mutation-bearing tumor cells (Fig. 3A). These results indicate that MsMab-1 is useful in immunohistochemical analyses for the detection of IDH1-R132S/R132G mutations.
Discussion
Two novel drugs against mutated IDH1 and mutated IDH2 were recently developed. One inhibitor of mutant IDH1 (AGI-5198) blocked the ability of mutant IDH1 to produce 2-HG (Rohle et al. 2013). Under conditions of R-2HG inhibition, AGI-5198 induced demethylation of histone H3K9me3 and expression of genes associated with gliogenic differentiation. Blockade of mutant IDH1 impaired the growth of glioma cells possessing mutant IDH1, but not that of IDH1 wild-type glioma cells without appreciable changes in genome-wide DNA methylation. Another inhibitor of mutant IDH2 (AGI-6780) inhibits the tumor-associated mutant IDH2/R140Q potently and selectively (Wang et al. 2013). Treatment with AGI-6780 induced differentiation of TF-1 erythroleukemia and primary human AML cells in vitro. Therefore, it may become much more important for molecular targeted therapy to detect mutant IDH1/2 using anti-mutated IDH1/2 mAbs in immunohistochemistry. We previously established several anti-mutated IDH1/2 mAbs: HMab-1 against IDH1-R132H (Takano et al. 2012), SMab-1 against IDH1-R132S (Kaneko et al. 2011), GMab-r1 against IDH1-R132G (Kaneko et al. 2013a), KMab-1 against IDH2-R172K (Kaneko et al. 2013b), MMab-1 against IDH2-R172M (Kaneko et al. 2013b), and WMab-1 against IDH2-R172W (Kato and Kaneko 2013). In this study, we newly established a multi-specific anti-mutated IDH1/2 mAb (MsMab-1), which can be extremely useful for diagnosis of IDH1/2 mutation-bearing tumors. The discrepancy about MsMab-1 reactivity between ELISA (Table 1) and Western blot analyses (Figs. 1 and 2) is expected to be resolved in the near future.
Dinardo et al. (2013) recently reported that serum 2-HG levels predict IDH mutations and clinical outcome in AML. Their data confirm that serum measurement of an oncometabolite 2-HG provides useful diagnostic and prognostic information. The combination of immunohistochemistry using our developed anti-mutated IDH mAbs and serum measurement of an oncometabolite 2-HG is expected to improve patient selection for IDH1-targeted therapy.
In conclusion, the newly established anti-IDH1/2 mutation-specific mAb, MsMab-1, is expected to be extremely useful for diagnosis and biological evaluation of IDH1/2 mutation-bearing tumors. Because other IDH1/2 mutations such as IDH1-R132V (von Deimling et al. 2011), IDH2-R172S (Sonoda et al. 2009; Pansuriya et al. 2011), IDH2-R172G (Yan et al. 2009; Pansuriya et al. 2011), and IDH2-R140Q (Paschka et al. 2010) have been reported in gliomas, AML, or cartilaginous tumors, the cross-reaction of MsMab-1 with those IDH1/2 mutations might increase the diagnostic rate in immunohistochemistry.
Acknowledgements
We thank Shingo Takano (University of Tsukuba) and Atsushi Natsume (Nagoya University) for providing us glioma specimens. This work was supported in part by the Platform for Drug Discovery, Informatics, and Structural Life Science (M.K.K., Y.K.), by Regional Innovation Strategy Support Program (Y.K.), and by KAKENHI (25462242, 23790185, 23701043): a Grant-in-Aid-for Scientific Research (C) (Y.K.) and Grants-in-Aid for Young Scientists (B) (M.K.K., Y.K.) from the Ministry of Education, Culture, Sports, Science and Technology of Japan, and by Intelligent Cosmos Academic Foundation (Y.K.).
Conflict of Interest
The authors have no conflict of interest to declare.
References
-
Amary,
M.F.,
Damato,
S.,
Halai,
D.,
Eskandarpour,
M.,
Berisha,
F.,
Bonar,
F.,
McCarthy,
S.,
Fantin,
V.R.,
Straley,
K.S.,
Lobo,
S.,
Aston,
W.,
Green,
C.L.,
Gale,
R.E.,
Tirabosco,
R.,
Futreal,
A.,
Campbell,
P.,
Presneau,
N. &
Flanagan,
A.M.
(2011) Ollier disease and Maffucci syndrome are caused by somatic mosaic mutations of IDH1 and IDH2. Nat. Genet., 43, 1262-1265.
-
Balss,
J.,
Meyer,
J.,
Mueller,
W.,
Korshunov,
A.,
Hartmann,
C. &
von Deimling,
A.
(2008) Analysis of the IDH1 codon 132 mutation in brain tumors. Acta Neuropathol., 116, 597-602.
-
Capper,
D.,
Zentgraf,
H.,
Balss,
J.,
Hartmann,
C. &
von Deimling,
A.
(2009) Monoclonal antibody specific for IDH1 R132H mutation. Acta Neuropathol., 118, 599-601.
-
Dang,
L.,
White,
D.W.,
Gross,
S.,
Bennett,
B.D.,
Bittinger,
M.A.,
Driggers,
E.M.,
Fantin,
V.R.,
Jang,
H.G.,
Jin,
S.,
Keenan,
M.C.,
Marks,
K.M.,
Prins,
R.M.,
Ward,
P.S.,
Yen,
K.E.,
Liau,
L.M.,
Rabinowitz,
J.D.,
Cantley,
L.C.,
Thompson,
C.B.,
Vander Heiden,
M.G. &
Su,
S.M.
(2009) Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature, 462, 739-744.
-
Dinardo,
C.D.,
Propert,
K.J.,
Loren,
A.W.,
Paietta,
E.,
Sun,
Z.,
Levine,
R.L.,
Straley,
K.S.,
Yen,
K.,
Patel,
J.P.,
Agresta,
S.,
Abdel-Wahab,
O.,
Perl,
A.E.,
Litzow,
M.R.,
Rowe,
J.M.,
Lazarus,
H.M.,
Fernandez,
H.F.,
Margolis,
D.J.,
Tallman,
M.S.,
Luger,
S.M. &
Carroll,
M.
(2013) Serum 2-hydroxyglutarate levels predict isocitrate dehydrogenase mutations and clinical outcome in acute myeloid leukemia. Blood, 121, 4917-4924.
-
Hartmann,
C.,
Meyer,
J.,
Balss,
J.,
Capper,
D.,
Mueller,
W.,
Christians,
A.,
Felsberg,
J.,
Wolter,
M.,
Mawrin,
C.,
Wick,
W.,
Weller,
M.,
Herold-Mende,
C.,
Unterberg,
A.,
Jeuken,
J.W.,
Wesseling,
P.,
Reifenberger,
G. &
von Deimling,
A.
(2009) Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: a study of 1,010 diffuse gliomas. Acta Neuropathol., 118, 469-474.
-
Ikota,
H.,
Nobusawa,
S.,
Tanaka,
Y.,
Yokoo,
H. &
Nakazato,
Y.
(2011) High-throughput immunohistochemical profiling of primary brain tumors and non-neoplastic systemic organs with a specific antibody against the mutant isocitrate dehydrogenase 1 R132H protein. Brain Tumor Pathol., 28, 107-114.
-
Kaneko,
M.K.,
Tian,
W.,
Takano,
S.,
Suzuki,
H.,
Sawa,
Y.,
Hozumi,
Y.,
Goto,
K.,
Yamazaki,
K.,
Kitanaka,
C. &
Kato,
Y.
(2011) Establishment of a novel monoclonal antibody SMab-1 specific for IDH1-R132S mutation. Biochem. Biophys. Res. Commun., 406, 608-613.
-
Kaneko,
M.K.,
Tsujimoto,
Y.,
Hozumi,
Y.,
Goto,
K. &
Kato,
Y.
(2013a) Novel monoclonal antibodies GMab-r1 and LMab-1 specifically recognize IDH1-R132G and IDH1-R132L mutations. Monoclon. Antib. Immunodiagn. Immunother., 32, 224-228.
-
Kaneko,
M.K.,
Morita,
S.,
Tsujimoto,
Y.,
Yanagiya,
R.,
Nasu,
K.,
Sasaki,
H.,
Hozumi,
Y.,
Goto,
K.,
Natsume,
A.,
Watanabe,
M.,
Kumabe,
T.,
Takano,
S. &
Kato,
Y.
(2013b) Establishment of novel monoclonal antibodies KMab-1 and MMab-1 specific for IDH2 mutations. Biochem. Biophys. Res. Commun., 432, 40-45.
-
Kato,
Y.,
Jin,
G.,
Kuan,
C.T.,
McLendon,
R.E.,
Yan,
H. &
Bigner,
D.D.
(2009) A monoclonal antibody IMab-1 specifically recognizes IDH1R132H, the most common glioma-derived mutation. Biochem. Biophys. Res. Commun., 390, 547-551.
-
Kato,
Y. &
Kaneko,
M.K.
(2013) Generation of a novel monoclonal antibody WMab-1 specific for IDH2-R172W mutation. Biochem. Biophys. Res. Commun., 433, 374-378.
-
Kato,
Y.
Natsume,
A. &
Kaneko,
M.K.
(2013) A novel monoclonal antibody GMab-m1 specifically recognizes IDH1-R132G mutation. Biochem. Biophys. Res. Commun., 432, 564-567.
-
Kolker,
S.,
Pawlak,
V.,
Ahlemeyer,
B.,
Okun,
J.G.,
Horster,
F.,
Mayatepek,
E.,
Krieglstein,
J.,
Hoffmann,
G.F. &
Kohr,
G.
(2002) NMDA receptor activation and respiratory chain complex V inhibition contribute to neurodegeneration in d-2-hydroxyglutaric aciduria. Eur. J. Neurosci., 16, 21-28.
-
Pansuriya,
T.C.,
van Eijk,
R.,
d’Adamo,
P.,
van Ruler,
M.A.,
Kuijjer,
M.L.,
Oosting,
J.,
Cleton-Jansen,
A.M.,
Oosterwijk,
J.G.,
van Verbeke,
S.L.,
Meijer,
D.,
van Wezel,
T.,
Nord,
K.H.,
Sangiorgi,
L.,
Toker,
B.,
Liegl-Atzwanger,
B.,
San-Julian,
M.,
Sciot,
R.,
Limaye,
N.,
Kindblom,
L.G.,
Daugaard,
S.,
Godfraind,
C.,
Boon,
L.M.,
Vikkula,
M.,
Kurek,
K.C.,
Szuhai,
K.,
French,
P.J. &
Bovee,
J.V.
(2011) Somatic mosaic IDH1 and IDH2 mutations are associated with enchondroma and spindle cell hemangioma in Ollier disease and Maffucci syndrome. Nat. Genet., 43, 1256-1261.
-
Parsons,
D.W.,
Jones,
S.,
Zhang,
X.,
Lin,
J.C.,
Leary,
R.J.,
Angenendt,
P.,
Mankoo,
P.,
Carter,
H.,
Siu,
I.M.,
Gallia,
G.L.,
Olivi,
A.,
McLendon,
R.,
Rasheed,
B.A.,
Keir,
S.,
Nikolskaya,
T.,
Nikolsky,
Y.,
Busam,
D.A.,
Tekleab,
H.,
Diaz,
L.A. Jr.,
Hartigan,
J.,
Smith,
D.R.,
Strausberg,
R.L.,
Marie,
S.K.,
Shinjo,
S.M.,
Yan,
H.,
Riggins,
G.J.,
Bigner,
D.D.,
Karchin,
R.,
Papadopoulos,
N.,
Parmigiani,
G.,
Vogelstein,
B.,
Velculescu,
V.E. &
Kinzler,
K.W.
(2008) An integrated genomic analysis of human glioblastoma multiforme. Science, 321, 1807-1812.
-
Paschka,
P.,
Schlenk,
R.F.,
Gaidzik,
V.I.,
Habdank,
M.,
Kronke,
J.,
Bullinger,
L.,
Spath,
D.,
Kayser,
S.,
Zucknick,
M.,
Gotze,
K.,
Horst,
H.A.,
Germing,
U.,
Dohner,
H. &
Dohner,
K.
(2010) IDH1 and IDH2 mutations are frequent genetic alterations in acute myeloid leukemia and confer adverse prognosis in cytogenetically normal acute myeloid leukemia with NPM1 mutation without FLT3 internal tandem duplication. J. Clin. Oncol., 28, 3636-3643.
-
Rohle,
D.,
Popovici-Muller,
J.,
Palaskas,
N.,
Turcan,
S.,
Grommes,
C.,
Campos,
C.,
Tsoi,
J.,
Clark,
O.,
Oldrini,
B.,
Komisopoulou,
E.,
Kunii,
K.,
Pedraza,
A.,
Schalm,
S.,
Silverman,
L.,
Miller,
A.,
Wang,
F.,
Yang,
H.,
Chen,
Y.,
Kernytsky,
A.,
Rosenblum,
M.K.,
Liu,
W.,
Biller,
S.A.,
Su,
S.M.,
Brennan,
C.W.,
Chan,
T.A.,
Graeber,
T.G.,
Yen,
K.E. &
Mellinghoff,
I.K.
(2013) An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science, 340, 626-630.
-
Sonoda,
Y.,
Kumabe,
T.,
Nakamura,
T.,
Saito,
R.,
Kanamori,
M.,
Yamashita,
Y.,
Suzuki,
H. &
Tominaga,
T.
(2009) Analysis of IDH1 and IDH2 mutations in Japanese glioma patients. Cancer Sci., 100, 1996-1998.
-
Struys,
E.A.,
Salomons,
G.S.,
Achouri,
Y.,
Van Schaftingen,
E.,
Grosso,
S.,
Craigen,
W.J.,
Verhoeven,
N.M. &
Jakobs,
C.
(2005) Mutations in the D-2-hydroxyglutarate dehydrogenase gene cause D-2-hydroxyglutaric aciduria. Am. J. Hum. Genet., 76, 358-360.
-
Takano,
S.,
Kato,
Y.,
Yamamoto,
T.,
Kaneko,
M.K.,
Ishikawa,
E.,
Tsujimoto,
Y.,
Matsuda,
M.,
Nakai,
K.,
Yanagiya,
R.,
Morita,
S.,
Tsuboi,
K. &
Matsumura,
A.
(2012) Immunohistochemical detection of IDH1 mutation, p53, and internexin as prognostic factors of glial tumors. J. Neurooncol., 108, 361-373.
-
von Deimling,
A.,
Korshunov,
A. &
Hartmann,
C.
(2011) The next generation of glioma biomarkers: MGMT methylation, BRAF fusions and IDH1 mutations. Brain Pathol., 21, 74-87.
-
Wang,
F.,
Travins,
J.,
DeLaBarre,
B.,
Penard-Lacronique,
V.,
Schalm,
S.,
Hansen,
E.,
Straley,
K.,
Kernytsky,
A.,
Liu,
W.,
Gliser,
C.,
Yang,
H.,
Gross,
S.,
Artin,
E.,
Saada,
V.,
Mylonas,
E.,
Quivoron,
C.,
Popovici-Muller,
J.,
Saunders,
J.O.,
Salituro,
F.G.,
Yan,
S.,
Murray,
S.,
Wei,
W.,
Gao,
Y.,
Dang,
L.,
Dorsch,
M.,
Agresta,
S.,
Schenkein,
D.P.,
Biller,
S.A.,
Su,
S.M.,
de Botton,
S. &
Yen,
K.E.
(2013) Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science, 340, 622-626.
-
Ward,
P.S.,
Patel,
J.,
Wise,
D.R.,
Abdel-Wahab,
O.,
Bennett,
B.D.,
Coller,
H.A.,
Cross,
J.R.,
Fantin,
V.R.,
Hedvat,
C.V.,
Perl,
A.E.,
Rabinowitz,
J.D.,
Carroll,
M.,
Su,
S.M.,
Sharp,
K.A.,
Levine,
R.L. &
Thompson,
C.B.
(2010) The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell, 17, 225-234.
-
Yan,
H.,
Parsons,
D.W.,
Jin,
G.,
McLendon,
R.,
Rasheed,
B.A.,
Yuan,
W.,
Kos,
I.,
Batinic-Haberle,
I.,
Jones,
S.,
Riggins,
G.J.,
Friedman,
H.,
Friedman,
A.,
Reardon,
D.,
Herndon,
J.,
Kinzler,
K.W.,
Velculescu,
V.E.,
Vogelstein,
B. &
Bigner,
D.D.
(2009) IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med., 360, 765-773.