Review
Myofibroblasts: Biochemical and Proteomic Approaches to Fibrosis
2013 年 230 巻 2 号 p. 67-73
詳細
2013 年 230 巻 2 号 p. 67-73
Fibrosis is a state, in which excess amounts of extracellular matrix are deposited in the tissue. Fibrosis can occur in various organs, including the liver, lung, kidney and heart. The progression of fibrosis involves interstitial hypercellularity, accumulation of extracellular matrix, and atrophy of epithelial structures, resulting in a loss of normal function. Myofibroblasts play a crucial role in the development and progress of fibrosis. When stimulated, myofibroblasts actively synthesize connective tissue components and cause organ fibrosis. As a result, the process and the mechanism of myofibroblast activation represent a target for antifibrotic treatment. As yet, however, an effective treatment has not been developed, and new treatment modalities are expected. Because activation of myofibroblasts is a key event during fibrosis development, there is great interest in identifying and characterizing proteins whose expression is changed after this activation. In this review, fibrosis is outlined and the role of myofibroblasts in this disorder is described. Furthermore, the search for candidate proteins to target for treatment and the prospects of antifibrotic therapy are discussed.
Organ fibrosis is a clinical condition caused by an excessive deposition of extracellular matrix (ECM), which impairs organ function (MacKenna et al. 2000). Chronic tissue injury leads to fibrosis in many organs, including the liver, lung, kidney and heart. Fibrosis occurs when normal tissue remodeling fails to terminate. Tissue repair involves the ability of fibroblasts to synthesize and secrete components of the ECM. These responses are necessary for the normal wound healing process; however, this process occasionally fails to stop or an excessive response occurs. This enhanced reaction is the cause of fibrosis. Some fibrogenic responses to transforming growth factor-β (TGF-β), including α-smooth muscle actin (α-SMA) expression and ECM contraction, are hallmarks of the development of fibrosis (Santibañez et al. 2011).
Myofibroblasts are fibroblasts that express certain features of smooth muscle differentiation. They have contractility and are distributed over most internal organs. Activation of myofibroblasts is associated with morphological changes including cellular hypertrophy and the formation of actin stress fibers. During the process of wound healing in the skin, fibroblasts differentiate into myofibroblasts, thereby shrinking granulation tissue, such as scar and keloid tissue (Reisdorf et al. 2001; Zhou et al. 2010).
In the kidney, for example, mesangeal cells and tubular interstitial cells differentiate into myofibroblasts, and ultimately the kidney of an affected individual shows impaired function owing to glomerulosclerosis (Gerolymos et al. 2011). In the liver, hepatic stellate cells (also called Ito cells) differentiate into myofibroblasts and the subsequent deposition of ECM causes cirrhosis (Senoo et al. 2010). Granules containing vitamin A are present in stellate cells; after differentiation into myofibroblasts, however, these cells express α-SMA and do not contain vitamin A. The differentiated cells synthesize excess ECM proteins, leading to the development of liver cirrhosis.
Furthermore, after various stimuli in the lung, interstitial fibroblasts transdifferentiate into myofibroblasts, which synthesize and secrete collagen and other connective tissue proteins and cause pulmonary fibrosis (Goodwin and Jenkins 2009).
The accumulation of a large number of myofibroblasts is responsible for exaggerated and uncontrolled production of ECM during the development and progression of pathological fibrosis (Powell et al. 1999). As described above, myofibroblasts in fibrotic tissues are derived from fibroblasts, stellate cells, and mesangeal cells. Other sources of myofibroblasts are also known (Hinz et al. 2007). For example, epithelial-mesenchymal transition (EMT) is notable for its association with cancer metastasis (Tanjore et al. 2011). Bone marrow-derived circulating fibrocytes, pericytes, and cells derived from endothelial-mesenchymal transition (EndoMT) are also thought to be sources of myofibroblasts (Piera-Velazquez et al. 2011). The sources of myofibroblasts are summarized in Fig. 1.
In general, signal transduction plays an important role in the differentiation and/or activation of cells. Many signaling pathways are involved in myofibroblast activation (Fig. 2). Examples of signals that make myofibroblast activation are as follows. For example, TGF-β action is mediated through the TGF-β receptor (TGFR), which phosphorylates Smad; then phosphorylated Smad enters the nucleus, where it functions as a transcription factor. Connective tissue growth factor (CTGF) is thought to be a downstream mediator of TGF-β, with a particular role in stimulating fibroblast matrix production and myofibroblast differentiation (Scotton and Chambers 2007). Angiotensin II, a part of the renin-angiotensin system (RAS), causes vasoconstriction and an increase in raises blood pressure. Angiotensin II also acts as a potent growth factor and cytokine; its action is mediated through the angiotensin II receptor, which activates downstream signaling pathways (Yin et al. 2003). The mitogen-activated protein kinases (MAPKs) and phosphoinositide 3-kinase (PI3K) are central Toll-like receptor (TLR) signaling molecules (Li et al. 2008). Cannabinoids are derivatives of the marijuana components, and Cannabinoids exert their effects via CB1 and CB2 receptors. Nuclear factor erythroid 2-related factor 2 (Nrf2) is a transcription factor; Kelch-like ECH-associated protein 1 (Keap1) interacts with Nrf2 and acts as a negative regulator of Nrf2 (Motohashi and Yamamoto 2004). Some of the ligands and receptors of these intracellular signaling of these pathways are depicted in Fig. 2.
Activation of myofibroblasts by TGF-β plays a crucial role in the pathogenesis of human fibrotic diseases (Desmoulière 1995). When activated, myofibroblasts can secrete the following cytokines and chemokines: interleukin-1 (IL-1), IL-6, tumor necrosis factor α (TNF-α), IL-10 IL-8, monocyte chemotactic protein-1 (MCP-1), growth-related oncogene-1α (GRO-1α), macrophage inflammatory protein 1α (MIP-1α), MIP-2, regulated upon activation, normal T-cell expressed and secreted (RANTES) and neutrophil-activating protein-78 (ENA-78). A number of growth factors are also produced by activated myofibroblasts including TGF-β, colony stimulating factor-1 (CSF-1), platelet-derived growth factor-AA (PDGF-AA), PDGF-BB, basic fibroblast growth factor (bFGF), insulin-like growth factor I (IGF-I), nerve growth factor (NGF), keratinocyte growth factor (KGF), hepatocyte growth factor (HGF) and stem cell factor (SCF) (Wynn 2008; Baum and Duffy 2011).
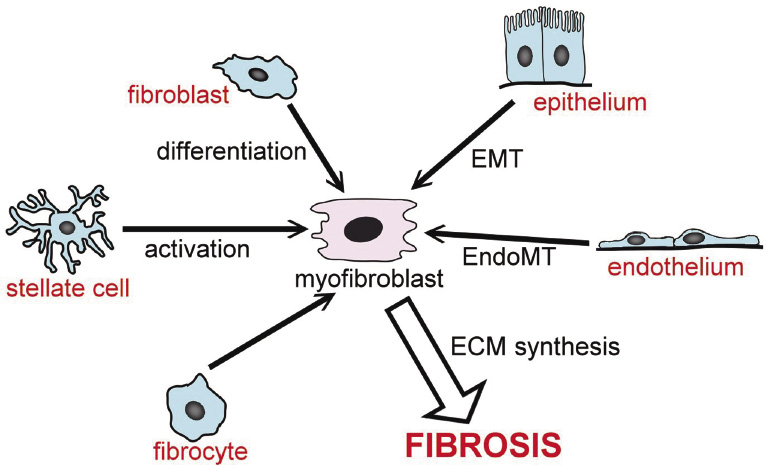
Origins of myofibroblasts. Various types of cell transdifferentiate into myofibroblasts. EMT, epithelial-to-mesenchymal transition. EndoMT, endothelial-to-mesenchymal transition. ECM, extracellular matrix.

Intracellular signaling pathways important in the excess deposition of extracellular matrix and fibrosis.
(A) TGF-β signaling begins on binding of TGF-β to the TGF-β type II receptor to form the type I/Type II receptor complex. Smad2/3 proteins are recruited to the receptor complex and released to interact with Smad4. TGF-β signals are also transmitted via Smad-independent pathways. (B) CTGF interacts with various receptors and enhances fibrosis. CTGF also binds to TGF-β and enhances its signal.
LRP, low density lipoprotein receptor-related protein; TrkA, tyrosine kinase receptor type A.
(C) Angiotensin I is converted to angiotensin II by ACE. Angiotensin II binds to AT1R to activate various signaling pathways.
ET-1, endothelin-1; PAI-1, plasminogen activator inhibitor-1; MCP-1, monocyte chemoattractant protein-1; VCAM-1, vascular cell adhesion molecule-1; ICAM-1, intercellular adhesion molecule-1.
(D) CB1 induces activation of the PI3K/PKB and ERK pathways. Conversely, CB2 decreases fibrotic changes via the effects of COX-2 and ROS by reducing myofibroblast accumulation.
PI3K, phosphatidyl inositol-3-kinase; PKB, protein kinase B; ERK, extracellular signal-regulated kinase; COX-2, cyclooxygenase-2; ROS, reactive oxygen species.
(E) TIRAP specifically mediates the Myd88-dependent pathway of TLR2 and TLR4.
TIRAP, TIR domain-containing adaptor protein; TIR, Toll/IL-1 receptor homology; Myd88, myeloid differentiation factor 88.
(F) Nrf2 activity is constitutively suppressed by Keap1. By stimulating ROS, Nrf2 escapes from the control of Keap1, enters into the nucleus, and activates the gene expression of antioxidant enzymes.
Myofibroblast activation is a key event in the development of fibrosis. Proteins that affect the activation process are potential targets for the treatment of fibrosis. The protein expression profile of the cells is largely changed by activation. Proteins that show increased expression after activation may also be candidates for therapeutic targets. Proteome analysis is a very effective means for comparing the protein expression profile before and after activation.
There have been many comparative proteomic studies to identify proteins that are differentially expressed between myofibroblasts and activated myofibroblasts. Many proteins in myofibroblasts whose expression is changed by activation of the cells have been identified by proteomic analysis as described below.
For example, it has been reported that PDGF stimulates the production of collagen VI, FK506-binding protein of 65 kDa, actin-related protein (ARP3), T-complex protein and heat shock protein 60 in myofibroblasts (Malmström et al. 2003). In addition, 89 differentially expressed proteins were identified by comparative proteome analysis of lung tissue from patients with sporadic idiopathic pulmonary fibrosis and tissue from control human donor lungs (Korfei et al. 2011). Furthermore, stellate cell activation-associated protein (STAP) was identified by proteome analysis of the activation of rat stellate cells (Kawada et al. 2001).
Proteome analysis of microdissected cirrhotic septa and liver parenchyma cells revealed increased expression of proteins associated with cell structure, including actin, prolyl 4-hydroxylase, tropomyosin, calponin, transgelin, and human microfibril-associated protein 4 (MFAP-4) in cirrhotic septa (Mölleken et al. 2009). In addition, in renal interstitial fibrosis caused by aristolochic acid administration to rats, upregulation of peroxiredoxin and downregulation of regucalcin and ATP synthase was reported (Wu et al. 2011).
We have also performed a proteome analysis using the cell line MRC-5, a myofibroblastic cell line (Jacobs et al. 1970) derived from fetal human lung fibroblasts. DIGE-MALDI-TOF analysis revealed that the expression of some actin-related proteins, molecular chaperones, and several enzymes was changed after treatment of the cells with TGF-β. With regard to actin-related proteins, transgelin (SM22), profilin 1, caldesmon, calpain small subunit 1, myosin IIIA, actin-related protein 2/3 complex subunit 2, actin-like protein 3, integrin α11, and tropomyosin β chain were identified. Among the molecular chaperones, glucose-regulated protein precursor, peroxyredoxin, heat-shock protein beta1, peptidyl-prolyl cis-trans isomerase and protein disulfide isomerase showed altered expression, as did the enzymes, glutathione S-transferase P, glyceraldehyde 3 phosphate dehydrogenase, ATP-dependent RNA helicase DDX1, malate dehydrogenase, alpha enolase, pyruvate kinase isozyme M1/M2, ATP synthase subunit alpha, creatine kinase, aldehyde dehydrogenase, and adenylate kinase. Moreover, it has been shown that treatment with TGF-β induces a set of actin-associated proteins that may contribute to the increased contractile properties of the myofibroblast (Malmström et al. 2004). Our own data, representative examples shown in Table 1, support these findings.
We also searched for proteins whose level of expression was reversely affected by activation and inactivation. In brief, we prepared TGF-β-stimulated MRC-5 cells and forskolin-treated MRC-5 cells and compared protein expression between these cells. As a result, tropomyosin β-chain, tropomyosin α-1 chain, myosin regulatory light polypeptide 2, caldesmon and transgelin were found to be increased by TGF-β treatment and decreased by forskolin treatment. The large majority of these proteins have been reported to be more or less associated with fibrosis. In particular, tropomyosin β-chain, tropomyosin α-1 chain, myosin regulatory light polypeptide 2, caldesmon and transgelin are components of the cytoskeleton, and it is likely and reasonable that the changes in expression of these proteins are associated with myofibroblast activation and inactivation.
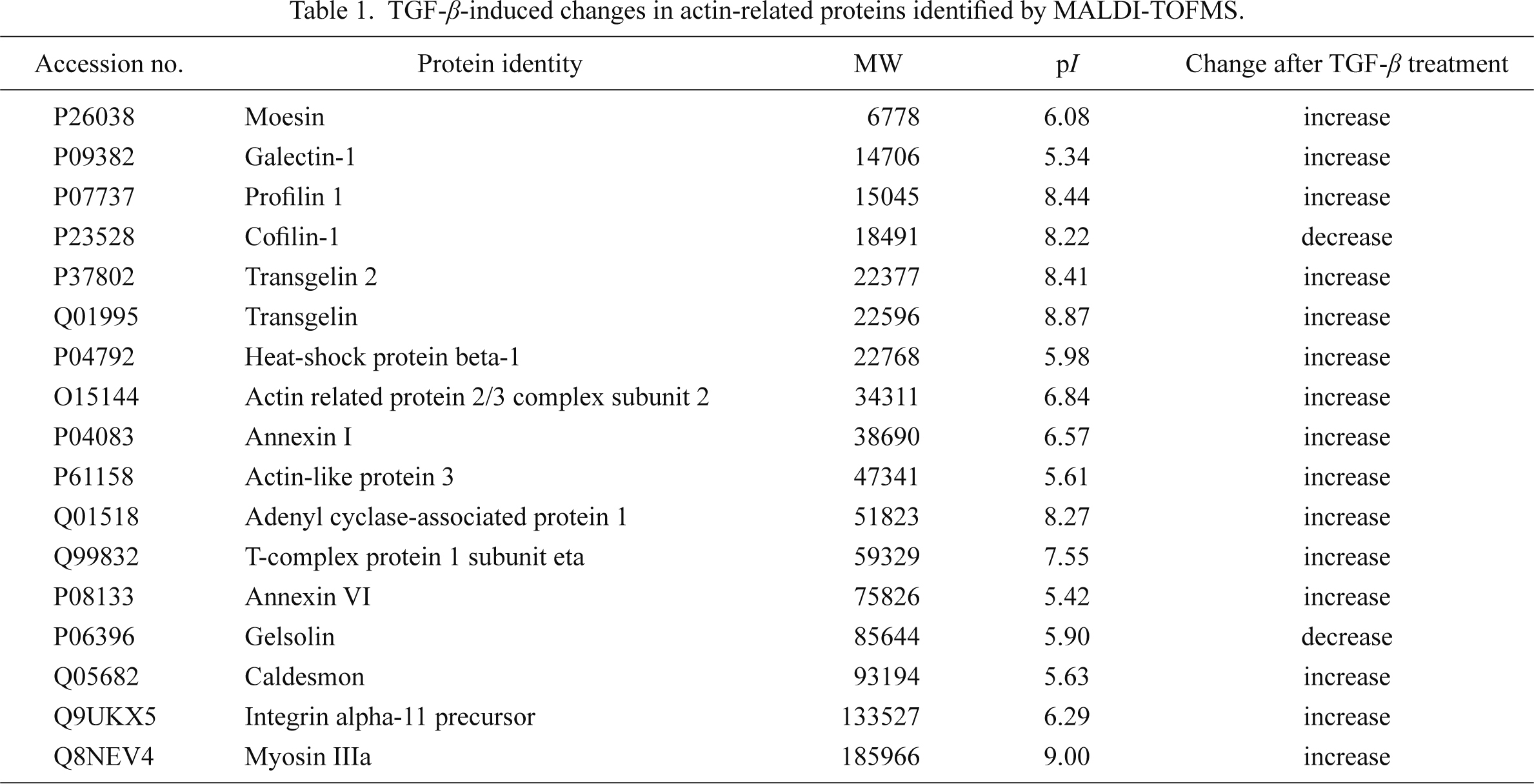
TGF-β-induced changes in actin-related proteins identified by MALDI-TOFMS.
Accession numbers are from the Swiss-Prot database.
TGF-β is a strong activator of myofibroblasts and is known to be a potent fibrotic growth factor. With regard to the fibrotic process, important actions of TGF-β are related to the deposition of ECM and to cell differentiation. TGF-β causes the deposition and remodeling of ECM by stimulating cells to simultaneously (i) increase the synthesis of most matrix proteins (Ihn 2002); (ii) decrease the production of matrix-degrading proteases and increase the production of inhibitors of these proteases (Knittel et al. 1999); and (iii) modulate the expression of integrins (Margadant and Sonnenberg 2010).
For the treatment of fibrosis, suppression of TGF-β activity has been tested in animal models; for example, administration of anti-TGF-β antibody was found to prevent glomerulonephritis in rat (Border et al. 1990). In addition, we reported that gene transfer of an anti-TGF-β reagent, decorin, prevents experimental pulmonary fibrosis (Shimizukawa et al. 2003). These studies have been successfully done with an experimental animal model but have not been applied to clinical cases, probably due to the fact that TGF-β is a multifunctional cytokine and inhibition of its activity would affect not only progression of fibrosis but also many important events in the body.
CTGF is a down-stream effector of TGF-β (Holmes et al. 2001) and represents a potential target for fibrosis therapy. We searched for proteins that interact with CTGF using a yeast two-hybrid system and found that fibronectin binds to this growth factor (Yoshida and Munakata 2007). The biological significance of this interaction is currently under investigation. TGF-β signaling is mediated by both Smad-dependent and Smad-independent signal transduction pathways, such as p38 MAPK. Both types of the pathways are targets for antifibrotic therapy (Wang et al. 2005; Li et al. 2008; Zhou et al. 2010).
Angiotensin II is a well-studied member of the RAS pathway and not only acts as a vasopressor but also potently activates myofibroblasts and induces the production of reactive oxygen species (ROS) (Rodríguez-Iturbe and García García 2010). There are many studies on the prevention of fibrosis by angiotensin converting enzyme inhibitors (ACEIs) and angiotensin II receptor blockers (ARBs) (Tsang et al. 2004). For example, we previously reported that treatment with ARBs and ACEI prevented the development of chronic pancreatitis and fibrosis in Wistar Bonn/Kobori rats (Sakurai et al. 2011). As mentioned above, angiotensin II also induces the production of ROS (Thakur et al. 2010; Barnes and Gorin 2011).
Generation of ROS has been linked to pulmonary fibrosis. It has been reported that ROS activate RhoA (Kondrikov et al. 2011), a factor that controls cell adhesion and motility via reorganization of the actin cytoskeleton and regulation of actin-myosin filament bundles. Rho-kinase is an effector of Rho, and the Rho/Rho-kinase pathway plays an important role in various cellular functions. It has been shown that the Rho/Rho-kinase pathway is involved in pulmonary fibrosis; thus, blocking this pathway may work as an antifibrotic therapy (Shimizu et al. 2001).
Nrf2 is a critical transcription factor responsible for antioxidant responsive element (ARE)-dependent transcription (Itoh et al. 1999; Ohtsuji et al. 2008), and many antioxidative enzymes are transcriptionally regulated through AREs in their promoter regions (Itoh et al. 2010). Recently, it was reported that nuclear translocation of Nrf2 plays a role in myofibroblast dedifferentiation (Artaud-Macari et al. 2013). It has been shown that subcellular localization of Nrf2 is under the control of Keap1 (Itoh et al. 2003; Watai et al. 2007). As a result, the Nrf2-Keap1 system is a potential candidate for antifibrotic therapy target.
Another candidate for antifibrotic therapy target is the cannabinoid receptor. The effects of cannabinoids are mediated by two different cannabinoid receptors, CB1 (Matsuda et al. 1990) and CB2 (Munro et al. 1993). It has been demonstrated that activation of hepatic cannabinoid CB2 receptor limits the progression of experimental liver fibrosis, and that CB1 receptor antagonists inhibit the progression of fibrosis (Julien et al. 2005; Teixeira-Clerc et al. 2006).
In addition, the TLR family comprises a number of conserved pattern recognition receptors. Upregulation of TLR2 during renal fibrosis development (Leemans et al. 2009) and the essential roles of TLR4 in both hepatic fibrosis (Seki et al. 2007) and lung fibrosis (Yoshizaki et al. 2008) have been reported. Thus, the TLR family could also be a target for antifibrotic therapy.
Activated myofibroblasts are the primary target for antifibrotic therapy, because a reduction in activated myofibroblasts may lead to a reversal of fibrosis (Brenner 2009; Kisseleva and Brenner 2011). Inactivation of myofibroblasts is also expected to cease fibrosis.
We recently observed that MRC-5 cells were inactivated after treatment with supernatant derived from a culture of A549 cells, a human lung carcinoma cell line (Park et al. 2012). Inactivation was accompanied by morphological changes in the MRC-5 cells and a decrease in α-SMA expression. We purified the inactivation factor from the culture supernatant by gel chromatography and reversed-phase chromatography. The substance was analyzed by GC-MS and NMR and identified as 5′-deoxy-5′-(methylthio) adenosine (MTA). The mechanism of inactivation by this factor was then investigated, which showed that MTA increased phosphorylation of cAMP response element binding protein (CREB). Furthermore, increased phosphorylation of extracellular signal-regulated kinase (ERK) 1/2 due to TGF-β in the cultured cells was blocked by MTA. ERK1/2 phosphorylation in lung fibrosis induced by bleomycin was also attenuated by MTA, showing that the effects of MTA on myofibroblast inactivation are related to CREB and ERK1/2 phosphorylation (Park et al. 2012).
Fibrosis occurs in various organs. The intractable diseases cirrhosis, renal and glomerular sclerosis, and interstitial pneumonitis are types of fibrosis occurring in the liver, kidney, and lung, respectively.
Myofibroblast activation plays a pivotal role in the pathogenesis of fibrosis. Changes associated with the activation of myofibroblasts have been examined in detail, and many approaches have been tried for the development of antifibrotic therapies. From recent advances in basic and clinical studies, promising candidates for antifibrotic therapy have been identified.
The authors declare no conflict of interest.