Abstract
The steroidogenic enzyme 21-hydroxylase is necessary for the synthesis of both glucocorticoids and mineralocorticoids. 21-hydroxylase is a cytochrome P-450 enzyme and is encoded by the gene CYP21A2. Here we report a 68-year-old phenotypically ‘male’ but genetically female patient with 21-hydroxylase deficiency (21OHD) and the concomitant virilizing adrenocortical carcinoma. This patient grew up as a male and has not encountered any episodes of adrenal insufficiency without glucocorticoid replacement in his lifetime. A chromosome test at admission, however, identified the 46, XX karyotype, and serum 17-hydroxyprogesterone and urine pregnanetriolone and 11β-hydroxyandrostendione were all elevated, consistent with 21OHD. Moreover, serum testosterone was 1.90 ng/ml, much higher than the female standard levels, and serum cortisol was 5.7 µg/ml, slightly lower than standard levels. Genetic analysis identified the patient as a heterozygote of the two pathogenic mutations in the CYP21A2 gene: IVS2-13C(A)>G and R356W. Magnetic resonance imaging (MRI) revealed the presence of left adrenal tumor measuring 6 cm, which was subsequently diagnosed as adrenocortical carcinoma based on the criteria of Weiss. Immunohistochemical analysis of the tumor specimens revealed the expression of various enzymes involved in testosterone production, including 3β-hydroxysteroid dehydrogenase, 17α-hydroxylase/17,20-lyase, and 17β-hydroxysteroid dehydrogenase. Importantly, the expression of immunoreactive 21-hydroxylase was detected in these tumor cells. The levels of adrenal tumor-derived steroid metabolites were all markedly decreased following the surgery. This is the first report on a virilized 21OHD patient associated with the adrenocortical tumor that produces testosterone. Moreover, the concomitant adrenocortical tumor may ameliorate adrenocortical insufficiency by producing cortisol.
Introduction
The steroidogenic enzyme 21-hydroxylase is necessary for the synthesis of both glucocorticoids and mineralocorticoids, and its deficiency is responsible for over 90% of the cases with congenital adrenal hyperplasia (CAH). The enzyme 21-hydroxylase is a cytochrome P-450 enzyme, also known as P450c21. The genes encoding P450c21, CYP21A2, and a pseudogene, CYP21P, are located on chromosome 6p21.3, adjacent to genes C4B and C4A, encoding two isoforms of the fourth component in the class III region of the HLA complex (Carroll et al. 1985; White et al. 1985). Mutations in the CYP21A2 gene usually result in various degrees of impaired cortisol and aldosterone synthesis and excess androgens (Merke and Bornstein 2005; Nimkarn et al. 2011). The clinical presentation of 21-hydroxylase deficiency (21OHD) has been further classified as the classical form, including the salt wasting (SW) and simple virilizing (SV) subclasses and the non-classical (NC) or late-onset form, based upon the different degrees of deficiency in enzymatic activity (Zhang et al. 2009). In Japan, a nationwide neonatal mass-screening program for CAH measuring serum 17α-hydroxyprogesterone (17OHP) levels has been available since 1989, which has also made those with classical 21OHD diagnosed at an older age extremely unlikely (Shinagawa et al. 2007). In addition, the patients with 21OHD are associated with marked proliferation of the zonae fasciculate and reticularis especially in untreated conditions with excessive adrenocorticotropic hormone (ACTH) levels, but rarely developed adrenocortical carcinoma, except for a few cases of classical 21OHD (Hamwi et al. 1957; Bauman and Bauman 1982; Jaursch-Hancke et al. 1988). Virilized 46, XX classical 21OHD patients are rarely raised as male (Lee and Houk 2010).
In this report, we described a 68-year-old phenotypically male but genetically female classical 21OHD patient associated with testosterone production and adrenocortical carcinoma. The concomitant adrenocortical carcinoma may ameliorate adrenocortical insufficiency by producing cortisol.
Patient and Methods
Patient
A 68-year-old Japanese phenotypically male (Fig. 1A and B) presented with left adrenal incidentaloma, which was identified when he underwent abdominal computed tomography (CT) as the health check-up (Fig. 1C). The adrenal tumor measured 34.9 × 54.7 × 55.0 mm. The patient was raised as a man since birth but underwent testis plasty due to testicular dysgenesis when the patient was 11 years old but he has never been diagnosed as 21OHD at all until the current admission. Family history was not contributory. Physical examination on admission revealed that height was 138 cm, weight 51 kg, and body mass index 26. The pubic hair was kinky and thin but the patient was phenotypically male based on his muscular extremities and the resemblance of the external genitalia to a penis rather than a clitoris (Fig. 1A and B).
Laboratory data on first admission were summarized in Table 1. Adrenocortical hormonal abnormalities were clinically not apparent. Abdominal magnetic resonance imaging (MRI) demonstrated that the right adrenal gland was morphologically normal (Fig. 1D) with artificial materials in the scrotum (Fig. 1G). In addition, the internal genitalia simulating uterus or ovary was located in the space left dorsal to the urinary bladder, suggesting either anomalies of Mullerian duct or testis formation, respectively (Fig. 1E and F). Urine cortisol excretion was slightly below normal. The patient did not have any clinical symptoms suggestive of adrenocortical insufficiency such as general malaise, but primary adrenocortical insufficiency was hormonally suspected because of an increased ACTH level up to 110 pg/ml and a decreased cortisol level of 5.7 μg/ml (Table 1). The diagnosis was confirmed by subsequent ACTH test (see Fig. 2H). Oral administration of hydrocortisone (10 mg per day) was subsequently initiated to treat this adrenocortical insufficiency.
In addition to the findings above, the serum testosterone levels (1.90 ng/ml) of this phenotypically male but genetically female patient was found to be lower than the male (2.07-7.61) but higher than the female standard levels (0.13-0.69) (Table 1), as well as basal serum levels of luteinizing hormone (LH; < 0.01 mIU/ml) and follicle-stimulating hormone (FSH; 1.48 mIU/ml). These data were consistent with the presence of hypogonadotropic hypogonadism, consistent with the results of the LH-releasing hormone (LHRH)-stimulation test (Fig. 2A and B). In addition, ACTH response to corticotropin releasing hormone (CRH) was exaggerated (Fig. 2F). Other pituitary functions were within normal limits (Fig. 2C-E).
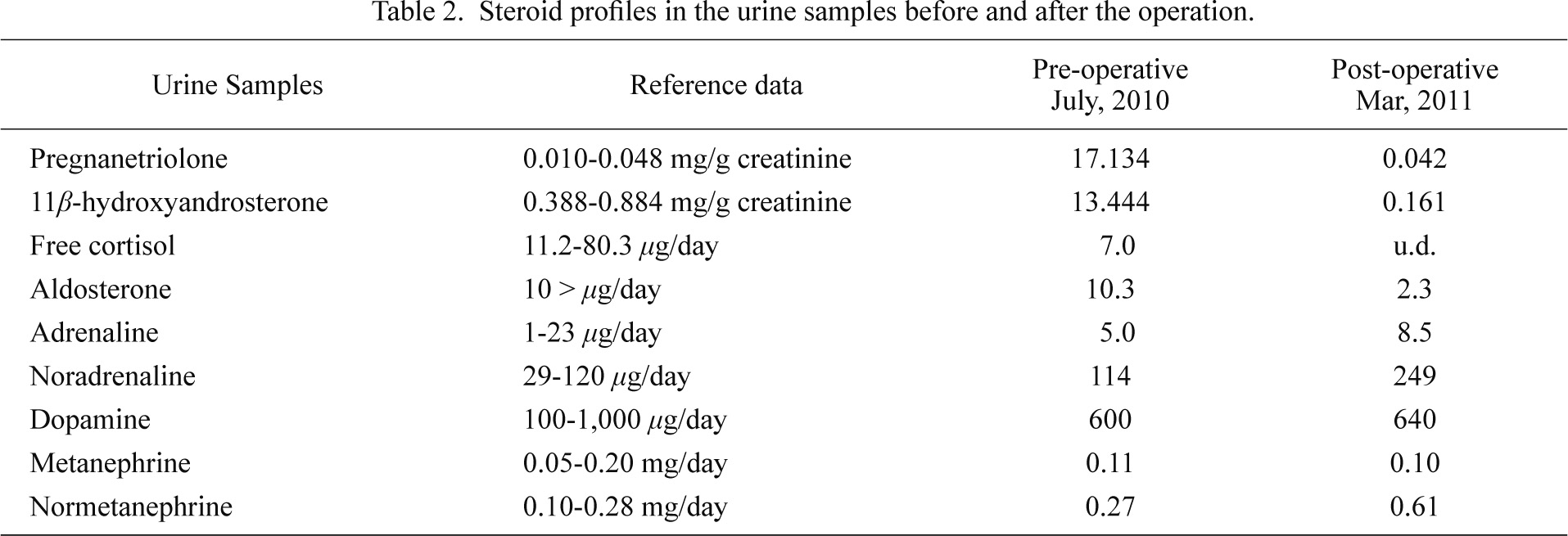
Due to the possibility of malignancy based on the tumor size (diameter 6 cm), the patient underwent a laparoscopic left adrenalectomy in August 2010. After the operation, hydrocortisone was tapered. When the dose was reduced to 10 mg/day, which was the same dose used before the operation, the patient clinically experienced bouts of marked general fatigue consistent with an increased serum ACTH level and decreased serum cortisol and aldosterone levels with 10 mg hydrocortisone administration. Progression of adrenal insufficiency was suspected, and hydrocortisone (20 mg/day) was orally re-administered. Re-evaluation of both pituitary and adrenal functions in March 2011 revealed the normalization of serum LH (20.71 mIU/ml) and FSH (57.49 mIU/ml) levels, while LHRH-stimulated LH and FSH responses had recovered but were not completely normalized (Fig. 2A and B). In contrast, serum testosterone level had markedly declined to undetectable ones (0.05 > ng/ml) and serum estradiol level also decreased from 28 pg/ml to undetectable levels (10 > pg/ml) after the operation. We recommended testosterone supplementation to this phenotypically male but genetically female patient to maintain masculinization but the patient eventually declined the hormone therapy. The external penis-like genitalia became more atrophic at this visit compared to before the operation. CRH-stimulated serum ACTH and cortisol responses were excessive and mild, respectively (Fig. 2F and G). Post-operative basal and ACTH-stimulated serum cortisol levels were lower than pre-operative levels (Fig. 2H). Results of other pituitary function tests were the same as before the surgery (Fig. 2C-E). Urine free-cortisol and aldosterone were decreased to undetectable levels (Table 2). These findings all indicated that the primary adrenal insufficiency had progressed following the resection of the adrenal tumor in this patient despite the hydrocortisone replacement therapy.
Blood samples were collected for genetic analysis after appropriate written informed consent had been obtained in accordance with the Declaration of Helsinki. After the diagnosis, the patient received psychological counseling by his doctor.
Immunohistochemical analysis of steroidogenic enzymes was performed on 10% formalin-fixed paraffin-embedded specimens employing the biotin-strept avidin (B-SA) amplified method using the StrAvi-Gen B-SA immunostaining system (Biogenex, CA, USA). The antibodies used were against 21-hydroxylase (CYP21) (White et al. 1984), 17α-hydroxylase/17,20-lyase (CYP17) (Sasano et al. 1992), dehydroepiandrosterone sulfotransferase (DHEAST) (Santa Cruz, CA, USA), 3β-hydroxysteroid dehydrogenase (3βHSD) (Lorence et al. 1990), or 17β-hydroxysteroid dehydrogenase type 5 (17βHSD5) (Sigma-Aldrich, MO, USA) (Nakamura et al. 2009). For positive control of immunostaining, we used normal human adrenal (CYP21, CYP17, DHEAST and 3βHSD) and testis (17βHSD5). For negative control, we used normal rabbit IgG (CYP21, CYP17, DHEAST and 3βHSD) or 0.01M PBS (17βHSD5) instead of the primary antibodies and no specific immunoreactivity was detected (data not shown). To further determine the characteristics of the adrenal tumor, monoclonal mouse Ki-67 and CD31 antibodies (DAKO, Denmark), and monoclonal mouse CD34 antibody (Nichirei, Tokyo, Japan) were used for immunochemistry. Immunostaining procedures and properties of primary antibodies used in this study have been described in detail previously (Sasano et al. 1992).
Direct sequence analysis
Genomic DNA from blood samples was subjected to direct sequencing of the coding exons and flanking splice sites of CYP21A2, using previously reported polymerase chain reaction (PCR) primers (Koyama et al. 2002). To confirm a heterozygous mutation, the corresponding PCR products were subcloned with a TOPO TA Cloning Kit (Invitrogen, Carlsbad, CA, USA), and the two alleles were sequenced separately.
Urine steroid profile analysis
Urine samples were randomly collected for the study and kept at −20°C until analysis. We studied this patient and control women (n = 19, 50-59 years of age) for urinary steroid profile by gas chromatography-mass spectrometry/selected ion monitoring (GC-MS-SIM), as reported (Homma et al. 2003; Koyama et al. 2012). In brief, 0.05- to 0.2- mL urine samples were subjected to enzymatic hydrolysis, organic solvent extraction and methyloximetrimethylsilyl derivatization, and the derivative was subjected to GC-MS-SIM analysis. GC-MS-SIM analysis was performed on an HP5890II GC with an HP-Ultra1 fused silica column (25 m × 0.2 nm × 0.33 μm) coupled to an HP5973MS (Agilent Technologies, Santa Clara, CA, USA).
Results
Genetic analysis
A chromosome test identified the 46, XX karyotype by both G-banding and fluorescent in-situ hybridization (FISH), and the expression of the sex determination gene on chromosome Y (SRY) was negative on chromosome X, indicating that the patient was female and not the typical XX male. Genetic analysis by PCR-direct sequencing disclosed two mutations in the CYP21A2 gene: IVS2-13C(A)>G in intron 2 and R356W in exon 8. Of these, IVS2-13C(A)>G is a substitution of a nucleotide at the 13th position from the splice donor site of exon 3 (C or A in unaffected individuals) to G. This mutation has been shown to cause aberrant splicing and subsequent frame-shift, leading to a premature stop codon (Higashi et al. 1988). R356W is a missense mutation that substitutes Trp for Arg356 in exon 8 (Fig. 3) (Koyama et al. 2002).
Serum and Urine steroid profile
A serum steroid profile performed to rule out CAH, as shown in Table 1, showed an extraordinarily high level of serum 17-hydroxyprogesterone 17OHP indicating 21OHD. A urine steroid profile showed that pregnanetriolone (a metabolite of 21-deoxycortisol) and 11β-hydroxyandrosterone (a metabolite of 11β-hydroxyandrostenedione), both sensitive indicators of 21OHD (Homma et al. 2004; Koyama et al. 2012), were higher than reference levels (Table 2), confirming the diagnosis of 21OHD. At birth, the patient did not have a SW crisis, and was sufficiently masculinized to be male phenotype despite the 46, XX karyotype. Based on all these findings, we diagnosed this patient with 21OHD, classical SV type.
Macroscopic and histopathological findings of the adrenal tumor
The tumor weighed 60 g and its outer surface appeared yellow to brown but the cut surface appeared black or dark brown (Fig. 1H), presumably derived from accumulated lipofuscin in the cytosol of tumor cells. Normal adrenal was attached to the tumor (data not shown). We evaluated the cell proliferation by determining a Ki-67 labeling index of 4.5% (Iino et al. 1997; Nakazumi et al. 1998) (Fig. 4A). Histopathological examination also demonstrated the presence of venous invasion demonstrated by CD31 immunostaining (Fig. 4B), and sinusoidal invasion by CD34 immunostaining (Fig. 4C). The tumor was subsequently diagnosed as adrenocortical carcinoma according to the Weiss’s criteria (four <sinusoidal invasion, venous invasion, cytoplasm and capsular invasion> of nine criteria were met) of adrenocortical malignancy (Weiss 1984).
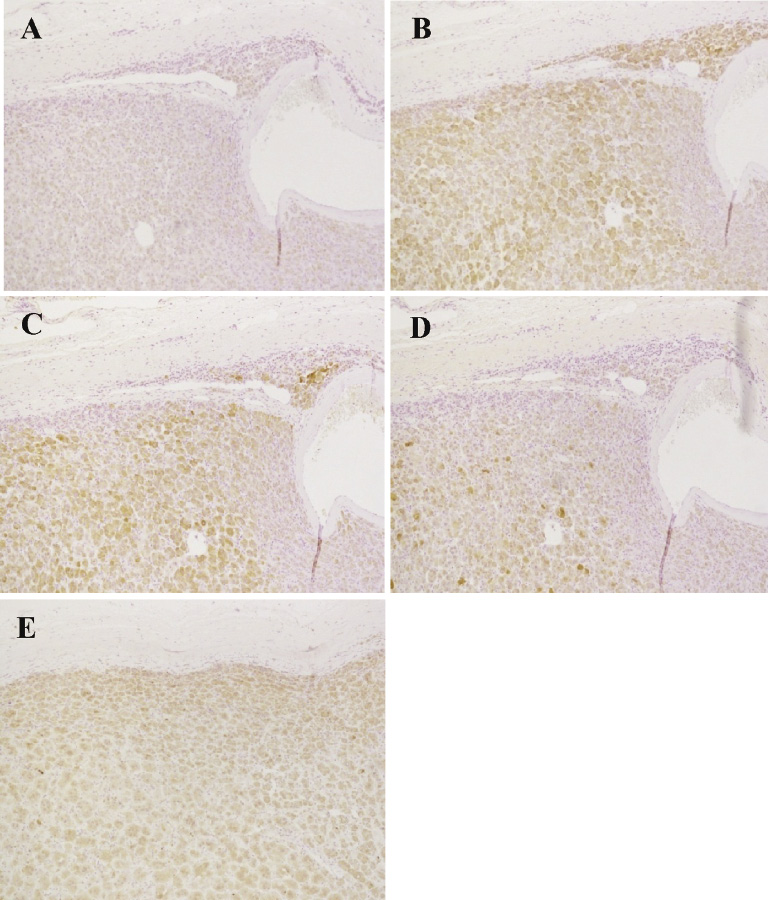
Immunohistochemistry of steroidogenic enzymes revealed weak but discernible immunoreactivity of CYP21 in the tumor (Fig. 5A), and immunoreactive 3βHSD, CYP17, and DHEAST were all clearly detected (Fig. 5B-D). No positive cells were detected with control IgG (data not shown). We also demonstrated the presence of 17βHSD5, a testosterone biosynthesis enzyme (Fig. 5E) in the tumor tissue, which accounted for the increased testosterone levels in this patient before the surgery.
Discussion
In this study, we demonstrated the presence of 21OHD, classic SV type and testosterone-producing adrenocortical carcinoma in the 68-year-old phenotypically male but genetically female patient with treatment naïve. This rather complicated patient is considered unique and informative because of the following three clinical and hormonal aspects: (1) the adrenocortical tumor turned out to be histopathologically malignant or carcinoma, (2) the tumor was considered to produce testosterone as a result of ectopic expression of 17βHSD5 as well as other corticosteroids, which contribute to the prevention of adrenocortical insufficiency despite no cortisol replacement and possibly further masculinization, and (3) unique patterns of 21OHD-related mutations, i.e., R356W and IVS2-13C(A)>G, both of which have been reported more frequently in SW type than in SV type.
The prevalence of adrenal incidentaloma among 21OHD patients appear to be relatively high both in heterozygous and homozygous carriers of CAH (Jaresch et al. 1992). However, it is also true that relatively few cases of adrenocortical carcinoma have been reported in the literature in the patients with CAH (Hamwi et al. 1957; Bauman and Bauman 1982; Jaursch-Hancke et al. 1988). One similar patient has been reported, in which 21OHD, classical SV type, presented as massive adrenal masses in the seventh decade of life (Abo et al. 1999).
In our present patient, 21OHD was not diagnosed at birth, but firstly identified at the age of 68 years old. Among the patients with CAH, complete virilization, such as the male external genitalia with absent testis, is well-known but actually rare and almost all (~95%) 46, XX CAH infants develop a female phenotype (Lee and Houk 2010). However, the present patient was apparently male and the sexual identity of this particular patient had been male for the last 67 years. The prominent masculinization in this patient might have been accelerated by the existence of an adrenal tumor in addition to the phenotype of the classical SV type at birth. One possible explanation for this marked masculinization since birth was that the hormone-producing tumor existed at an earlier stage of life but gradually transformed from benign to malignant at some point, although the process of adrenal tumorigenesis has remained unclear. 17βHSD5, which regulates the conversion of androstenedione to testosterone, was expressed in human adrenal cortex, as judged by immunohistochemical analysis (Nakamura et al. 2009). However, 17βHSD5 mRNA level in the adrenals were much lower than that in the liver and prostate (Dufort et al. 1999). Therefore, the 17βHSD5 immunoreactivity in the tumor tissue of this patient is considered as an ectopic expression and to contribute to the overproduction of testosterone. Serum testosterone levels were higher than those in females, suggesting that increased testosterone levels might have contributed to the further masculinization before or after adolescence, although the period when the tumor developed remains unknown.
Post-operative urine cortisol levels decreased compared to the pre-operative levels, which suggested that cortisol was indeed produced by this concomitant carcinoma of this patient. In addition, the patient developed symptoms of adrenal insufficiency which progressed after adrenal tumor resection, although no symptoms prior to the operation despite no glucocorticoid replacement therapy. These findings all indicated that the adrenal tumor produced at least the steroid hormones required for maintaining water and mineral balance, essential for supporting life. The production of cortisol by the tumor is supported by the finding that the ectopic but discernible expression of CYP21 in the tumor. To the best of our knowledge, there is one report that suggests the production of cortisol by the adrenal tumor arising in the patient with 21OHD (Nagasaka et al. 1996). They reported that adrenal insufficiency developed after the surgical removal of the adrenal tumor and the pathological diagnosis was adrenocortical adenoma. Therefore, these tumors arising in the patients with 21OHD are considered to produce cortisol and contribute to the prevention of adrenocortical insufficiency despite no cortisol replacement through ectopic expression of CYP21. Preoperative levels of pregnanetriolone and 11β-hydroxyandrosterone were higher than post-operative levels and steroid hormones derived from the adrenal gland (except for orally administered hydrocortisone metabolites) were also decreased after the operation, indicating that the right adrenal gland did not produce enough steroid hormones, although it awaits further investigations for clarification. Of interest, the steroid production of left adrenal tumor was not necessarily autonomous based on the results of the 1 mg dexamethasone suppression test (Table 1). The fact that suppressed LH and FSH secretion recovered following the operation might be due to the interference of excessive androstenedione and hypersecretion of progesterone with LH and FSH secretion (Bachelot et al. 2008) but it also awaits further investigations for clarification.
In our patient, we identified the two distinct mutations of R356W and IVS2-13C(A)>G in the CYP21A2 gene. R356W completely abolishes the enzyme activity of 21-hydroxylase, and IVS2-13C(A)>G results in the decrease or loss of the enzyme activity due to a frame-shift (Wedell et al. 1994; Koyama et al. 2002). Therefore, it is surprising that the present patient with classical SV type did not develop adrenal insufficiency. In fact, the numbers of SW/SV/NC forms harboring the present mutations were reported to be 3/1/0 (Koyama et al. 2002), 3/0/0 (Stikkelbroeck et al. 2003), 1/0/0 (Usui et al. 2004), 4/1/0 (Wilson et al. 1995), and 3/0/0 (Krone et al. 2000), respectively. In addition, to the best of our knowledge, this is the first report that shows the patient with classical SV type associated with adrenocortical carcinoma harboring the above double mutations.
In summary, we report a 68-year-old phenotypically male but genetically female patient with 21OHD, classical SV type, accompanied by unilateral adrenocortical carcinoma. Incidentally, the concomitant adrenocortical carcinoma produced steroid hormones, such as cortisol and testosterone, thereby contributing to the prevention of adrenocortical insufficiency and contributing to masculinization.
Funding
This study did not receive any specific grant from any funding agency in the public, commercial, or not-for-profit sector.
Conflict of Interest
The authors have no conflict of interest.
References
-
Abo,
K.,
Sumino,
K.,
Nishio,
H.,
Hozumi,
T.,
Ishida,
Y.,
Fujieda,
K.,
Tajima,
T. &
Kazumi,
T.
(1999) 21-Hydroxylase deficiency presenting as massive bilateral adrenal masses in the seventh decade of life. Endocr. J., 46, 817-823.
-
Bachelot,
A.,
Chakthoura,
Z.,
Rouxel,
A.,
Dulon,
J. &
Touraine,
P.
(2008) Classical forms of congenital adrenal hyperplasia due to 21-hydroxylase deficiency in adults. Horm. Res., 69, 203-211.
-
Bauman,
A. &
Bauman,
C.G.
(1982) Virilizing adrenocortical carcinoma. Development in a patient with salt-losing congenital adrenal hyperplasia. JAMA, 248, 3140-3141.
-
Carroll,
M.C.,
Campbell,
R.D. &
Porter,
R.R.
(1985) Mapping of steroid 21-hydroxylase genes adjacent to complement component C4 genes in HLA, the major histocompatibility complex in man. Proc. Natl. Acad. Sci. USA, 82, 521-525.
-
Dufort,
I.,
Rheault,
P.,
Huang,
X.F.,
Soucy,
P. &
Luu-The,
V.
(1999) Characteristics of a highly labile human type 5 17beta-hydroxysteroid dehydrogenase. Endocrinology, 140, 568-574.
-
Hamwi,
G.J.,
Serbin,
R.A. &
Kruger,
F.A.
(1957) Does adrenocortical hyperplasia result in adrenocortical carcinoma. N. Engl. J. Med., 257, 1153-1157.
-
Higashi,
Y.,
Tanae,
A.,
Inoue,
H.,
Hiromasa,
T. &
Fujii-Kuriyama,
Y.
(1988) Aberrant splicing and missense mutations cause steroid 21-hydroxylase [P-450(C21)] deficiency in humans: possible gene conversion products. Proc. Natl. Acad. Sci. USA, 85, 7486-7490.
-
Homma,
K.,
Hasegawa,
T.,
Masumoto,
M.,
Takeshita,
E.,
Watanabe,
K.,
Chiba,
H.,
Kurosawa,
T.,
Takahashi,
T. &
Matsuo,
N.
(2003) Reference values for urinary steroids in Japanese newborn infants: gas chromatography/mass spectrometry in selected ion monitoring. Endocr. J., 50, 783-792.
-
Homma,
K.,
Hasegawa,
T.,
Takeshita,
E.,
Watanabe,
K.,
Anzo,
M.,
Toyoura,
T.,
Jinno,
K.,
Ohashi,
T.,
Hamajima,
T.,
Takahashi,
Y.,
Takahashi,
T. &
Matsuo,
N.
(2004) Elevated urine pregnanetriolone definitively establishes the diagnosis of classical 21-hydroxylase deficiency in term and preterm neonates. J. Clin. Endocrinol. Metab., 89, 6087-6091.
-
Iino,
K.,
Sasano,
H.,
Yabuki,
N.,
Oki,
Y.,
Kikuchi,
A.,
Yoshimi,
T. &
Nagura,
H.
(1997) DNA topoisomerase II alpha and Ki-67 in human adrenocortical neoplasms: a possible marker of differentiation between adenomas and carcinomas. Mod. Pathol., 10, 901-907.
-
Jaresch,
S.,
Kornely,
E.,
Kley,
H.K. &
Schlaghecke,
R.
(1992) Adrenal incidentaloma and patients with homozygous or heterozygous congenital adrenal hyperplasia. J. Clin. Endocrinol. Metab., 74, 685-689.
-
Jaursch-Hancke,
C.,
Allolio,
B.,
Metzler,
U.,
Bidlingmaier,
F. &
Winkelmann,
W.
(1988) Adrenocortical carcinoma in a patient with untreated congenital adrenal hyperplasia (CAH). Acta Endocrinologica (Copenh), 117, Suppl., 287, 146-147.
-
Koyama,
S.,
Toyoura,
T.,
Saisho,
S.,
Shimozawa,
K. &
Yata,
J.
(2002) Genetic analysis of Japanese patients with 21-hydroxylase deficiency: identification of a patient with a new mutation of a homozygous deletion of adenine at codon 246 and patients without demonstrable mutations within the structural gene for CYP21. J. Clin. Endocrinol. Metab., 87, 2668-2673.
-
Koyama,
Y.,
Homma,
K.,
Fukami,
M.,
Miwa,
M.,
Ikeda,
K.,
Ogata,
T.,
Hasegawa,
T. &
Murata,
M.
(2012) Two-step biochemical differential diagnosis of classic 21-hydroxylase deficiency and cytochrome P450 oxidoreductase deficiency in Japanese infants by GC-MS measurement of urinary pregnanetriolone/ tetrahydroxycortisone ratio and 11β-hydroxyandrosterone. Clin. Chem., 58, 741-747.
-
Krone,
N.,
Braun,
A.,
Roscher,
A.A.,
Knorr,
D. &
Schwarz,
H.P.
(2000) Predicting phenotype in steroid 21-hydroxylase deficiency? Comprehensive genotyping in 155 unrelated, well defined patients from southern Germany. J. Clin. Endocrinol. Metab., 85, 1059-1065
-
Lee,
P.A. &
Houk,
C.P.
(2010) Review of outcome information in 46,XX patients with congenital adrenal hyperplasia assigned/reared male: what does it Say about gender assignment? Int. J. Pediatr. Endocrinol., 2010, 982025.
-
Lorence,
M.C.,
Murry,
B.A.,
Trant,
J.M. &
Mason,
J.I.
(1990) Human 3 beta-hydroxysteroid dehydrogenase/delta 5—4isomerase from placenta: expression in nonsteroidogenic cells of a protein that catalyzes the dehydrogenation/isomerization of C21 and C19 steroids. Endocrinology, 126, 2493-2498.
-
Merke,
D.P. &
Bornstein,
S.R.
(2005) Congenital adrenal hyperplasia. Lancet, 365, 2125-2136.
-
Nagasaka,
S.,
Kubota,
K.,
Motegi,
T.,
Hayashi,
E.,
Ohta,
M.,
Takahashi,
K.,
Takahashi,
T.,
Iwasaki,
Y.,
Koike,
M. &
Nishikawa,
T.
(1996) A case of silent 21-hydroxylase deficiency with persistent adrenal insufficiency after removal of an adrenal incidentaloma. Clin. Endocrinol. (Oxf.), 44, 111-116.
-
Nakamura,
Y.,
Hornsby,
P.J.,
Casson,
P.,
Morimoto,
R.,
Satoh,
F.,
Xing,
Y.,
Kennedy,
M.R.,
Sasano,
H. &
Rainey,
W.E.
(2009) Type 5 17beta-hydroxysteroid dehydrogenase (AKR1C3) contributes to testosterone production in the adrenal reticularis. J. Clin. Endocrinol. Metab., 94, 2192-2198.
-
Nakazumi,
H.,
Sasano,
H.,
Iino,
K.,
Ohashi,
Y. &
Orikasa,
S.
(1998) Expression of cell cycle inhibitor p27 and Ki-67 in human adrenocortical neoplasms. Mod. Pathol., 11, 1165-1170.
-
Nimkarn,
S.,
Lin-Su,
K. &
New,
M.I.
(2011) Steroid 21 hydroxylase deficiency congenital adrenal hyperplasia. Pediatr. Clin. North Am., 58, 1281-1300, xii.
-
Sasano,
H.,
Miyazaki,
S.,
Sawai,
T.,
Sasano,
N.,
Nagura,
H.,
Funahashi,
H.,
Aiba,
M. &
Demura,
H.
(1992) Primary pigmented nodular adrenocortical disease (PPNAD): immunohistochemical and in situ hybridization analysis of steroidogenic enzymes in eight cases. Mod. Pathol., 5, 23-29.
-
Shinagawa,
T.,
Horikawa,
R.,
Isojima,
T.,
Naiki,
Y.,
Tanaka,
T. &
Katsumata,
N.
(2007) Nonclassic steroid 21-hydroxylase deficiency due to a homozygous V281L mutation in CYP21A2 detected by the neonatal mass-screening program in Japan. Endocr. J., 54, 1021-1025.
-
Stikkelbroeck,
N.M.,
Hoefsloot,
L.H.,
de Wijs,
I.J.,
Otten,
B.J.,
Hermus,
A.R. &
Sistermans,
E.A.
(2003) CYP21 gene mutation analysis in 198 patients with 21-hydroxylase deficiency in The Netherlands: six novel mutations and a specific cluster of four mutations. J. Clin. Endocrinol. Metab., 88, 3852-3859.
-
Usui,
T.,
Nishisho,
K.,
Kaji,
M.,
Ikuno,
N.,
Yorifuji,
T.,
Yasuda,
T.,
Kuzuya,
H. &
Shimatsu,
A.
(2004) Three novel mutations in Japanese patients with 21-hydroxylase deficiency. Horm. Res., 61, 126-132.
-
Wedell,
A.,
Thilen,
A.,
Ritzen,
E.M.,
Stengler,
B. &
Luthman,
H.
(1994) Mutational spectrum of the steroid 21-hydroxylase gene in Sweden: implications for genetic diagnosis and association with disease manifestation. J. Clin. Endocrinol. Metab., 78, 1145-1152.
-
Weiss,
L.M.
(1984) Comparative histologic study of 43 metastasizing and nonmetastasizing adrenocortical tumors. Am. J. Surg. Pathol., 8, 163-169.
-
White,
P.C.,
Grossberger,
D.,
Onufer,
B.J.,
Chaplin,
D.D.,
New,
M.I.,
Dupont,
B. &
Strominger,
J.L.
(1985) Two genes encoding steroid 21-hydroxylase are located near the genes encoding the fourth component of complement in man. Proc. Natl. Acad. Sci. USA, 82, 1089-1093.
-
White,
P.C.,
New,
M.I. &
Dupont,
B.
(1984) Cloning and expression of cDNA encoding a bovine adrenal cytochrome P-450 specific for steroid 21-hydroxylation. Proc. Natl. Acad. Sci. USA, 81, 1986-1990.
-
Wilson,
R.C.,
Mercado,
A.B.,
Cheng,
K.C. &
New,
M.I.
(1995) Steroid 21-hydroxylase deficiency: genotype may not predict phenotype. J. Clin. Endocrinol. Metab., 80, 2322-2329.
-
Zhang,
H.J.,
Yang,
J.,
Zhang,
M.N.,
Zhang,
W.,
Liu,
J.M.,
Wang,
W.Q.,
Ning,
G. &
Li,
X.Y.
(2009) Variations in the promoter of CYP21A2 gene identified in a Chinese patient with simple virilizing form of 21-hydroxylase deficiency. Clin. Endocrinol. (Oxf.), 70, 201-207.