Abstract
Nucleotide excision repair (NER) is an essential biological pathway protecting against ultraviolet light-induced DNA damage. Deficient NER causes a group of rare genetic disorders including two autosomal recessive diseases, xeroderma pigmentosum (XP) and Cockayne syndrome (CS). In addition to the cutaneous photosensitivity shared in XP and CS, CS is featured by growth failure, neurological deterioration, microcephaly, and deep sunken eyes. XP/CS complex is an extremely rare type of NER disorder with a distinct phenotype that is characterized by the skin and eye pathology of XP and the somatic and neurological abnormalities of CS. Some of CS cases have been reported to be complicated with renal failure, but the genetic background or the etiology of the renal failure has not been reported. We herein report a 1-year-old Japanese boy with XP/CS complex, complicated by nephrotic syndrome. Diagnosis was confirmed by the presence of compound heterozygous mutations, G47R (c.139G>A) and R616G (c.1846C>G), in the excision repair cross-complementation group 2 (ERCC2) gene. The kidney biopsies, performed at the age of 1 year and 2 months, revealed diffuse expansion of the mesangial matrix and segmental glomerulosclerosis under light microscopy, and diffused thin capillary walls with partially lamellated regions under electron microscopy. Notably, high levels of urinary 8-hydroxy-2′-deoxyguanosin, known as an oxidative stress marker, were observed during the clinical course. The patient died at the age of 1 year and 11 months because of renal failure. We suggest the involvement of oxidative stress in the pathogenesis of nephrotic syndrome in NER disorders.
Introduction
Nucleotide excision repair (NER) is an essential biological pathway to protect against ultraviolet (UV) light-induced DNA damage in living organisms (Moriwaki 2015). Deficient NER causes rare genetic disorders including xeroderma pigmentosum (XP) and Cockayne syndrome (CS) (Kraemer et al. 2007: Moriwaki 2015). XP is an autosomal recessive disorder, and the XP patients are unable to repair DNA damage caused by UV light from the sun, thus, demonstrating markedly increased skin sun sensitivity. CS is a rare autosomal recessive disorder of NER pathway characterized by severe growth failure, progressive neurologic dysfunction, microcephaly, cutaneous photosensitivity, and a characteristic face with deep sunken eyes. CS has a broad phenotypic spectrum including CS type I, CS type II, and CS type III, which are classified based on age of onset, symptom severity, and rate of progression (Laugel 2013). XP/CS complex is known to have a distinct phenotype that is characterized by the skin and eye pathology of XP and the somatic and neurological abnormalities of CS (Broughton et al. 1995: Lehmann 2001: Fujimoto et al. 2005: Kraemer et al. 2007: Schäfer et al. 2013).
The NER pathway constitutes of dozens of genes and the products of these genes function sequentially in living organisms (Moriwaki 2015). Abnormality in one gene causes dysfunction of the subsequent steps in the NER pathway (Kraemer et al. 2007: Moriwaki 2015). Thus, there is a complex relationship between the clinical diseases and the molecular defects in NER diseases. Mutations in two major genes, excision repair cross-complementation group 6 (ERCC6) and ERCC8, have been identified as responsible for CS, and 65% and 35% of CS cases have mutations in ERCC6 and ERCC8, respectively (Kraemer et al. 2007: Suhasini and Brosh 2013). Mutations in three genes, ERCC2, ERCC3, and ERCC5, can be responsible for the XP/CS complex, on the other hand, mutation in each gene of them can also cause other types of NER diseases (Kraemer et al. 2007: Suhasini and Brosh 2013). Mutations in ERCC3 may cause XP and trichothiodystrophy (TTD), and mutations in ERCC5 may cause XP and cerebrooculofacioskeletal syndrome (COFS). Furthermore, mutation of ERCC2 can cause all three type of NER diseases, XP, TTD, and COFS.
Recent nationwide survey of CS in Japan has revealed that features such as body weight and height stagnation, language delay, abnormal nutritional pathways (tube feeding), and renal failure was more prominent in the deceased CS patients than surviving CS patients and about half of the deceased cases developed severe renal failure during the terminal stages of their condition (Kubota et al. 2015). This suggests that the prognosis of CS depends on the management of renal failure throughout the long-term clinical course. There are some reports describing CS patients with complications such as renal failure, but their molecular backgrounds have not been described in their case studies (Ohno and Hirooka 1966: Higginbottom et al. 1979: Hirooka et al. 1988: Sato et al. 1988: Reiss et al. 1996: Funaki et al. 2006).
In this article, we report the case study of a 1-year-old Japanese child with XP/CS complex caused by compound heterozygous mutations in the ERCC2 gene. The patient presented with an infantile onset of nephrotic syndrome and the cause of death was renal failure. The clinical course, renal pathology obtained by biopsy, and genetic background of the patient are herein presented. In addition, the amount of urinary 8-hydroxy-2′-deoxyguanosine (8-OHdG), which is known as a marker of oxidative stress in vivo, was determined in the clinical course of the disease in the patient.
Case Presentation
The patient was born to healthy non-consanguineous Japanese parents at 37 weeks by normal vaginal delivery, weighing 1,886 g. He showed failure to thrive and developmental delay at the age of 4 months. The symptoms of proteinuria and severe skin photosensitivity were manifested at the age of 10 months and the patient was admitted to our hospital at the age of 1 year.
On admission, his weight, height, and head circumference were 7.4 kg (−2.2 SD), 69 cm (−3.0 SD), and 36.8 cm (−6.0 SD), respectively. Physical characteristics of the patient included microcephaly, short neck, deep sunken eyes, short palpebral fissures, coarse hair, small penis, and bilateral undescended testes. The patient dermatologically showed depigmented macule, freckled face, and pigmentation. Neurologically, the patient showed spastic quadriplegia, uncontrolled head, and nystagmus. Prominent general edema and respiratory wheezing were also prominent on admission. Ophthalmological findings of the patient were microphthalmia, retinal atrophy, and hypoplasia of macula. Brain CT and MRI studies showed bilateral cortical calcifications and brain atrophy in the patient. All these findings suggested the diagnosis of XP/CS complex.
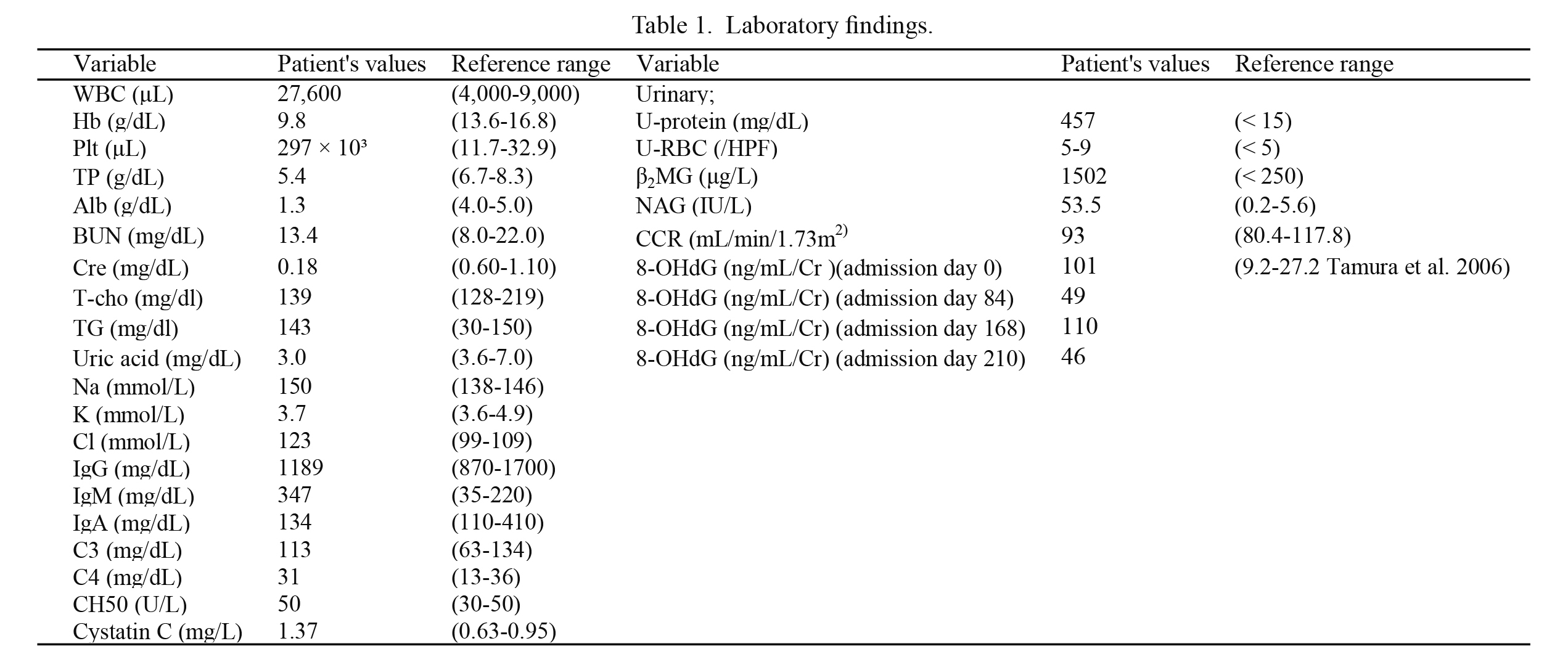
Ultrasound and CT studies showed no morphological abnormalities of the kidneys, and there was no history of urinary tract infection. No uptake was observed in the kidneys on Ga (Gallium-67) scintigraphy. Laboratory data showed hypoalbuminemia, hypernatremia, renal failure, proteinuria, and renal tubular damage. These laboratory findings suggesting renal dysfunction led us to perform renal biopsy to decide on course of treatment at the age of 1 year and 2 months (Table 1). A total of 28 glomeruli were histologically observed, showing global sclerosis in 2 glomeruli and diffuse expansion of the mesangial matrix and segmental sclerosis in the remaining 26 glomeruli (Fig. 1A). Electron microscopy revealed diffused thin capillary walls with partially lamellated regions (Fig. 1B).
DNA repair and genetic analyses and urinary 8-OHdG
As a diagnosis of CS was suspected in the patient, post-UV DNA repair and molecular studies using skin fibroblasts were performed. DNA repair tests, such as the measurement of post-UV unscheduled DNA synthesis and post-UV cell survival, were performed using the methods previously described (Moriwaki et al. 1996). These methods can help identify the gene responsible for the disease manifestation in the patient among the 6 genes, XPA, XPB, XPC, XPD, XPF, and XPG, which are known to cause 8 DNA repair diseases. This experiment suggested that XPD is responsible for XP/CS complex in the patient. A genetic analysis of the ERCC2 gene (XPD) was performed in the patient for confirmatory diagnosis of XP/CS complex caused by XPD. Genomic DNA was extracted from blood leukocytes and amplification of 23 exons and the exon-intron boundaries of the ERCC2 (NM_000400.3) was performed using polymerase chain reaction (PCR). The PCR products were directly sequenced in both the directions. This study was approved by the Ethics Committee of Akita University Graduate School of Medicine, and written informed consent was obtained from the subject’s guardians. The results showed that the patient had compound heterozygous mutations of G47R (c.139G>A) and R616G (c.1846C>G) (Fig. 2), leading to a diagnosis of XP/CS complex caused by XPD gene abnormality. Urinary 8-OHdG was determined using an enzyme immunoassay method at 4 time points in the clinical course of the patient. In all samples, the urinary 8-OHdG levels were increased, such as 101, 49, 110, and 46 ng/mL/Cr (Table 1). The normal range of urinary 8-OHdG depends on age in children and is reported as from 9.2 to 27.2 ng/mL/Cr in children between the age of 1 and 6 years (Tamura et al. 2006). The patient died at the age of 1 year and 11 months because of renal failure.

Discussion
We herein present a case of XP/CS complex complicated with renal failure clinically presenting nephrotic syndrome in infancy. The patient had compound heterozygous mutations of G47R (c.139G>A) and R616G (c.1846C>G) in the ERCC2 gene. The XPD protein encoded in the ERCC2 gene has two functions, NER and basal transcription (Taylor et al. 1997: Dubaele et al. 2003). In NER, the damaged DNA is cleaved after the damaged site is opened out by the helicase activity of XPD protein. The XPD protein is also a subunit of the transcription factor TFIIH complex, which has two quite separate roles in NER and in basal transcription by RNA polymerase II. The G47R, one mutant allele in the patient, is a previously known mutation of the ERCC2 that causes abolishment of both ATPase and DNA helicase activity of the XPD protein (Dubaele et al. 2003). The R616G, the other mutant allele in the patient, has not been reported before, but two missense mutations, R616P and R616W, have been reported at the same amino acid residue in the C-terminus region of XPD protein (Dubaele et al. 2003). These two mutations were analyzed by protein expression studies, demonstrating that mutations in the C-terminus of XPD protein prevent interaction with p44, which is another component of the transcription factor TFIIH. Thus, the R616G mutation is pathogenic and is a previously unreported mutation of the ERCC2 gene. The effect of the R616G was also predicted to be deleterious by in silico algorithms; Polyphen2 (http://genetics.bwh.harvard.edu/pph2), SIFT (http://sift.bii.a-star.edu.sg/), and Mutation Taster (http://www.mutationtaster.org/).
Nephrotic syndrome has been previously reported in several children with CS (Ohno and Hirooka 1966: Higginbottom et al. 1979: Hirooka et al. 1988: Sato et al. 1988: Reiss et al. 1996: Funaki et al. 2006). All these children share the same pathological features of biopsied renal tissues, including thickening of the glomerular basement membrane and mesangium, collapse of capillary loops, hyalinization of glomeruli, tubular atrophy, and interstitial fibrosis. Our patient was the youngest ever as a child of CS showing nephrotic syndrome, but the renal pathology of our case was compatible with the previous reports of those in CS. There results suggest that these patients share the same pathogenic background causing nephrotic syndrome.
We, for the first time, have measured urinary 8-OHdG in a CS patient, showing high level of urinary 8-OHdG, 101 ng/mL/Cr, in an acute phase of nephrotic syndrome. The levels of urinary 8-OHdG were high, such as 49, 110, and 46 ng/mL/Cr, during his treatment. 8-OHdG is formed from deoxyguanosine in DNA by hydroxyl free radicals and passes into the urine by glomerular filtration following the cleavage of specific enzymatic action. Elevations of urinary 8-OHdG have been reported in some diseases including type 1 diabetes and atopic dermatitis and proposed to be a stress marker in vivo in children (Tsukahara et al. 2003a, b). The 8-OHdG has also been evaluated as a marker of oxidative stress in patients with chronic renal failure previously (Akagi et al. 2003). Their results showed that serum 8-OHdG level was influenced by renal failure, but urinary 8-OHdG level was independent on renal function of patients. Thus, the high level of urinary 8-OHdG might show the existence of strong oxidative DNA damage in our patient, suggesting the relationship of oxidative stress and pathogenesis of nephrotic syndrome. We suggest the use of 8-OHdG as a biomarker of oxidative stress in NER.
In summary, we report an infant with XP/CS complex, wherein the XP/CS complex was caused by compound heterozygous mutations, G47R and R616G, in the ERCC2 gene showing nephrotic syndrome, resulting in early death due to renal failure. The renal biopsy showed typical pathological findings of renal tissue in CS. The patient clinically showed high levels of urinary 8-OGdG, suggesting the relationship between oxidative stress and pathogenesis of nephrotic syndrome.
Conflict of Interest
The authors declare no conflict of interest.
References
-
Akagi,
S.,
Nagake,
Y.,
Kasahara,
J.,
Sarai,
A.,
Kihara,
T.,
Morimoto,
H.,
Yano,
A.,
Nakao,
K.,
Nanba,
K.,
Ichikawa,
H. &
Makino,
H.
(2003) Significance of 8-hydroxy-2′-deoxyguanosine levels in patients with chronic renal failure. Nephrology (Carlton), 8, 192-195.
-
Broughton,
B.C.,
Thompson,
A.F.,
Harcourt,
S.A.,
Vermeulen,
W.,
Hoeijmakers,
J.H.J.,
Botta,
E.,
Stefanini,
M.,
King,
M.D.,
Weber,
C.A.,
Cole,
J.,
Arlett,
C.F. &
Lehmann,
A.R.
(1995) Molecular and cellular analysis of the DNA repair defect in a patient in xeroderma pigmentosum complementation group D who has the clinical features of xeroderma pigmentosum and Cockayne syndrome. Am. J. Hum. Genet., 56, 167-174.
-
Dubaele,
S.,
Proietti De Santis,
L.,
Bienstock,
R.J.,
Keriel,
A.,
Stefanini,
M.,
Van Houten,
B. &
Egly,
J.M.
(2003) Basal transcription defect discriminates between xeroderma pigmentosum and trichothiodystrophy in XPD patients. Mol. Cell, 11, 1635-1646.
-
Fujimoto,
M.,
Leech,
S.N.,
Theron,
T.,
Mori,
M.,
Fawcett,
H.,
Botta,
E.,
Nozaki,
Y.,
Yamagata,
T.,
Moriwaki,
S.,
Stefanini,
M.,
Momoi,
M.Y.,
Nakagawa,
H.,
Shuster,
S.,
Moss,
C. &
Lehmann,
A.R.
(2005) Two new XPD patients compound heterozygous for the same mutation demonstrate diverse clinical features. J. Invest. Dermatol., 125, 86-92.
-
Funaki,
S.,
Takahashi,
S.,
Murakami,
H.,
Harada,
K. &
Kitamura,
H.
(2006) Cockayne syndrome with recurrent acute tubulointerstitial nephritis. Pathol. Int., 56, 678-682.
-
Higginbottom,
M.C.,
Griswold,
W.R.,
Jones,
K.L.,
Vasquez,
M.D.,
Mendoza,
S.A. &
Wilson,
C.B.
(1979) The Cockayne syndrome: an evaluation of hypertension and studies of renal pathology. Pediatrics, 64, 929-934.
-
Hirooka,
M.,
Hirota,
M. &
Kamada,
M.
(1988) Renal lesions in Cockayne syndrome. Pediatr. Nephrol., 2, 239-243.
-
Kraemer,
K.H.,
Patronas,
N.J.,
Schiffmann,
R.,
Brooks,
B.P.,
Tamura,
D. &
DiGiovanna,
J.J.
(2007) Xeroderma pigmentosum, trichothiodystrophy and Cockayne syndrome: a complex genotype-phenotype relationship. Nueroscience, 145, 1388-1396.
-
Kubota,
M.,
Ohta,
S.,
Ando,
A.,
Koyama,
A.,
Terashima,
H.,
Kashii,
H.,
Hoshino,
H.,
Sugita,
K. &
Hayashi,
M.
(2015) Nationwide survey of Cockayne syndrome in Japan: incidence, clinical course and prognosis. Pediatr. Int., 57, 339-347.
-
Laugel,
V.
(2013) Cockayne syndrome: the expanding clinical and mutational spectrum. Mech. Ageing Dev., 134, 161-170.
-
Lehmann,
A.R.
(2001) The xeroderma pigmentosum group D (XPD) gene: one gene, two functions, three diseases. Genes Dev., 15, 15-23.
-
Moriwaki,
S.
(2015) Human DNA repair disorders in dermatology: A historical perspective, current concepts and new insight. J. Dermatol. Sci., 81, 77-84.
-
Moriwaki,
S.,
Stefanini,
M.,
Lehmann,
A.R.,
Hoeijmakers,
J.H.,
Robbins,
J.H.,
Rapin,
I.,
Botta,
E.,
Tanganelli,
B.,
Vermeulen,
W.,
Broughton,
B.C. &
Kraemer,
K.H.
(1996) DNA repair and ultraviolet mutagenesis in cells from a new patient with xeroderma pigmentosum group G and Cockayne syndrome resemble xeroderma pigmentosum cells. J. Invest. Dermatol., 107, 647-653.
-
Ohno,
T. &
Hirooka,
M.
(1966) Renal lesions in Cockayne’s syndrome. Tohoku J. Exp. Med., 89, 151-166.
-
Reiss,
U.,
Hofweber,
K.,
Herterich,
R.,
Waldherr,
R.,
Bohnert,
E.,
Jung,
E. &
Schärer,
K.
(1996) Nephrotic syndrome, hypertension, and adrenal failure in atypical Cockayne syndrome. Pediatr. Nephrol., 10, 602-605.
-
Sato,
H.,
Saito,
T.,
Kurosawa,
K.,
Ootaka,
T.,
Furuyama,
T. &
Yoshinaga,
K.
(1988) Renal lesions in Cockayne’s syndrome. Clin. Nephrol., 29, 206-209.
-
Schäfer,
A.,
Gratchev,
A.,
Seebode,
C.,
Hofmann,
L.,
Schubert,
S.,
Laspe,
P.,
Apel,
A.,
Ohlenbusch,
A.,
Tzvetkov,
M.,
Weishaupt,
C.,
Oji,
V.,
Schön,
M.P. &
Emmert,
S.
(2013) Functional and molecular genetic analyses of nine newly identified XPD-deficient patients reveal a novel mutation resulting in TTD as well as in XP/CS complex phenotypes. Exp. Dermatol., 22, 486-489.
-
Suhasini,
A.N. &
Brosh,
R.M. Jr.
(2013) Disease-causing missense mutations in human DNA helicase disorders. Mutat. Res., 752, 138-152.
-
Tamura,
S.,
Tsukahara,
H.,
Ueno,
M.,
Maeda,
M.,
Kawakami,
H.,
Sekine,
K. &
Mayumi,
M.
(2006) Evaluation of a urinary multi-parameter biomarker set for oxidative stress in children, adolescents and young adults. Free Radic. Res., 40, 1198-1205.
-
Taylor,
E.M.,
Broughton,
B.C.,
Botta,
E.,
Stefanini,
M.,
Sarasin,
A.,
Jaspers,
N.G.,
Fawcett,
H.,
Harcourt,
S.A.,
Arlett,
C.F. &
Lehmann,
A.R.
(1997) Xeroderma pigmentosum and trichothiodystrophy are associated with different mutations in the XPD (ERCC2) repair/transcription gene. Proc. Natl. Acad. Sci. USA, 94, 8658-8663.
-
Tsukahara,
H.,
Sekine,
K.,
Uchiyama,
M.,
Kawakami,
H.,
Hata,
I.,
Todoroki,
Y.,
Hiraoka,
M.,
Kaji,
M.,
Yorifuji,
T.,
Momoi,
T.,
Yoshihara,
K.,
Beppu,
M. &
Mayumi,
M.
(2003a) Formation of advanced glycosylation end products and oxidative stress in young patients with type 1 diabetes. Pediatr. Res., 54, 419-424.
-
Tsukahara,
H.,
Shibata,
R.,
Ohta,
N.,
Sato,
S.,
Hiraoka,
M.,
Ito,
S.,
Noiri,
E. &
Mayumi,
M.
(2003b) High levels of urinary pentosidine, an advanced glycation end product, in children with acute exacerbation of atopic dermatitis: relationship with oxidative stress. Metabolism, 52, 1601-1605.