Abstract
Aberrant DNA methylation, which can be detected in circulating cell-free DNA (cfDNA), is one of the major epigenetic alterations in hepatocellular carcinoma (HCC). UBE2Q1, a putative member of the ubiquitin-conjugating enzyme family, might play substantial roles in tumorigenesis. However, the methylation status of the UBE2Q1 gene in HCC remains unknown. We aimed to determine the methylation status of the UBE2Q1 gene promoter and to evaluate its potential clinical significance for HCC detection. The methylation-specific polymerase chain reaction (MSP) assay was used to detect the UBE2Q1 gene methylation status in serum samples from 80 patients with hepatitis B virus (HBV)-related HCC, 40 patients with liver cirrhosis (LC), 40 patients with chronic hepatitis B (CHB), and 20 healthy controls (HCs). Significantly lower methylation frequencies were detected in HCC patients (33.75%) compared with LC patients (55.00%, p = 0.026) and CHB patients (60.00%, p = 0.006) and HCs (65.00%, p = 0.011). Hypomethylation of the UBE2Q1 gene was negatively associated with the tumor node metastasis stage (rs = −0.30, p = 0.008). The UBE2Q1 gene methylation status combined with alpha fetoprotein using cut-off points of 20, 200 and 400 ng/ml showed sensitivity and specificity values of 58.8% and 75.0%, 53.8% and 87.5%, and 37.5% and 88.7%, respectively, and yielded a significantly increased area under the ROC curve (0.720, 0.760 and 0.694, respectively) for discriminating HCC from LC and CHB. Our study results suggest that hypomethylation of the UBE2Q1 gene promoter is a potential biomarker for detecting HBV-associated HCC.
Introduction
Hepatocellular carcinoma (HCC) is the most common type of liver cancer; in fact, HCC is the sixth most frequent cancer worldwide and the second leading cause of cancer-related death (Laursen 2014). Infection with hepatitis B or C virus, alcohol addiction, nonalcoholic steatohepatitis and exposure to aflatoxin B1 are all associated with hepatocellular carcinogenesis (Forner et al. 2012), and in China, hepatitis B virus (HBV) infection is the main risk factor for HCC (El-Serag and Rudolph 2007). Although advances in diagnosis and treatment have been achieved, the mortality rate associated with this cancer remains high. Indeed, the five-year survival rate of HCC is approximately 5% (Mohamed et al. 2012), due to late diagnosis or recurrence and metastasis after resection (Villanueva et al. 2010). Therefore, it is necessary to explore the molecular pathophysiology of HCC and identify new biomarkers for early diagnosis and surveillance.
Similar to many other tumors, the development of HCC involves complex genetic and epigenetic alterations, mutations, altered molecular pathways and chromosomal aberrations (Lachenmayer et al. 2010; Yates and Campbell 2012). In particular, accumulating evidence indicates that epigenetic modifications play a crucial role in HCC development (Nishida and Goel 2011). DNA methylation, which usually occurs at cytosines adjacent to guanines (CpG dinucleotides) (Cokus et al. 2008), and can thus lead to alterations in gene expression profiles, is one type of epigenetic mechanism (Kim et al. 2005), and aberrant DNA methylation of specific genes is a frequent event in HCC (Um et al. 2011). Circulating cell-free DNA (cfDNA) is a type of extracellular DNA, and a fraction of this cfDNAs in cancer patients is derived from tumor because the genetic and/or epigenetic molecular alterations characteristic of the tumor are found in the plasma or serum cfDNAs (Lecomte et al. 2010). DNA methylation can be detected in cfDNA from dead cancer cells in bodily fluids (Jahr et al. 2001; Chan and Baylin 2012). Several genes, including TFPI2, CDH1 and ACP1, have demonstrated aberrant methylation in the serum or tissue of HCC patients and thus hold clinical potential for HCC diagnosis and prognosis (Sun et al. 2013; Dou et al. 2016; Qiu et al. 2016). Furthermore, demethylating agents might reverse abnormal methylation (Perret 2011) and might thus represent a potential epigenetic cancer treatment strategy (Venturelli et al. 2013). Therefore, further study of abnormal methylation could provide a new avenue for diagnosing and treating HCC.
In eukaryotic cells, the ubiquitin proteasome system (UPS) is important for protein degradation, and approximately 80-90% of intracellular proteins are degraded through the UPS (Chen and Dou 2010). The UPS consists of ubiquitin, ubiquitination enzymes, deubiquitination enzymes and proteasomes (Zhong and Huang 2016). Deregulation of the UPS might cause tumorigenesis, which is closely related to cell proliferation, apoptosis, migration and invasion (Ding et al. 2014). The ubiquitin-conjugating enzyme 2 Q1 (UBE2Q1) gene is located on chromosome 1q21.3 and encodes E2, a member of the family of ubiquitin-conjugating enzymes (Grzmil et al. 2013). Recent studies have revealed that UBE2Q1 is overexpressed in breast carcinoma, colorectal tumors and acute lymphoblastic leukemia (Seghatoleslam et al. 2012; Shafiee et al. 2013, 2015) and might play a role as an oncogene. Another study indicated that the UBE2Q2 gene promoter is unmethylated, whereas a higher level of the methylated allele of UBE2Q1 has been detected in colorectal cancer tumor tissues compared with adjacent normal tissues and non-malignant controls (Mokarram et al. 2015). However, the DNA methylation status of the UBE2Q1 gene during HCC development remains virtually unknown.
Therefore, in the present study, we performed methylation-specific PCR (MSP) to determine the methylation of the UBE2Q1 gene promoter and evaluate its potential clinical significance for HCC detection and prognosis.
Materials and Methods
Patients
Patients with HBV infection-related HCC were recruited to participate in this study. We also recruited patients with HBV infection-related liver cirrhosis (LC), chronic hepatitis B (CHB) and healthy controls (HCs) from the Department of Hepatology, Qilu Hospital of Shandong University, from July 2015 to July 2016, as experimental controls. The HCC patients were enrolled according to the diagnostic criteria delineated in the 2010 updated version of the American Association for the Study of Liver Diseases (AASLD) Practice Guidelines for the Management of HCC (Bruix and Sherman 2011). The diagnosis of CHB and LC was made according to the guidelines for the prevention and treatment of CHB (2010) in China (Chinese Society of Hepatology and Chinese Society of Infectious Diseases, Chinese Medical Association 2011). All patients with other cancers, concurrent infection with hepatitis C virus, hepatitis D virus, or HIV and any other type of liver disease, such as alcoholic liver diseases, autoimmune liver diseases, non-alcoholic fatty liver diseases and other causes of chronic liver diseases, were excluded. A serum sample was collected from each participant prior to treatment. The study protocol was approved by the Ethics Committee of Qilu Hospital of Shandong University. Prior to collection of the serum samples, informed consent was obtained from each participant.
Collection of clinical data
The serum samples were collected after the participants had fasted for 12 hours. The serum biochemical markers of liver function that were assessed in this study using an automatic biochemical analyzer (Cobas c311, Roche Diagnostic Ltd., Germany) included alanine aminotransferase (ALT), aspartate aminotransferase (AST), total bilirubin (TBIl) and albumin (ALB). Prothrombin time activity (PTA) was evaluated with an ACL TOP 700 (Instrument Laboratory, USA). Hepatitis B e antigen (HBeAg) and alpha fetoprotein (AFP) were measured with an automatic biochemical analyzer (Roche Diagnostic Ltd., Germany).
All patients with HCC were diagnosed through an enhanced computed tomography (CT) scan (Discovery CT750, GE, USA), and the CT films were examined by a senior radiologist at the Department of Medical Imaging, Qilu Hospital, who was blinded to the patients’ characteristics. If the CT result was not typical, a liver biopsy was performed, and the diagnosis of HCC was confirmed by histology.
DNA extraction and sodium bisulfite modification
The total genomic DNA from 400 µl of sera was extracted using a QIAamp DNA Blood Mini Kit (Qiagen, GmbH, Hilden, Germany) according to the manufacturer’s instructions, and then stored at −20°C prior to sodium bisulfite modification.
Approximately 20 µl of the extracted DNA was subjected to bisulfite modification treatment using an EZ DNA Methylation-Gold KitTM (Zymo Research Corp, Orange, CA, USA) to convert all unmethylated cytosines to thymines. A final volume of 20 µl of modified DNA was stored at −20°C for further analysis.
Methylation-specific PCR (MSP)
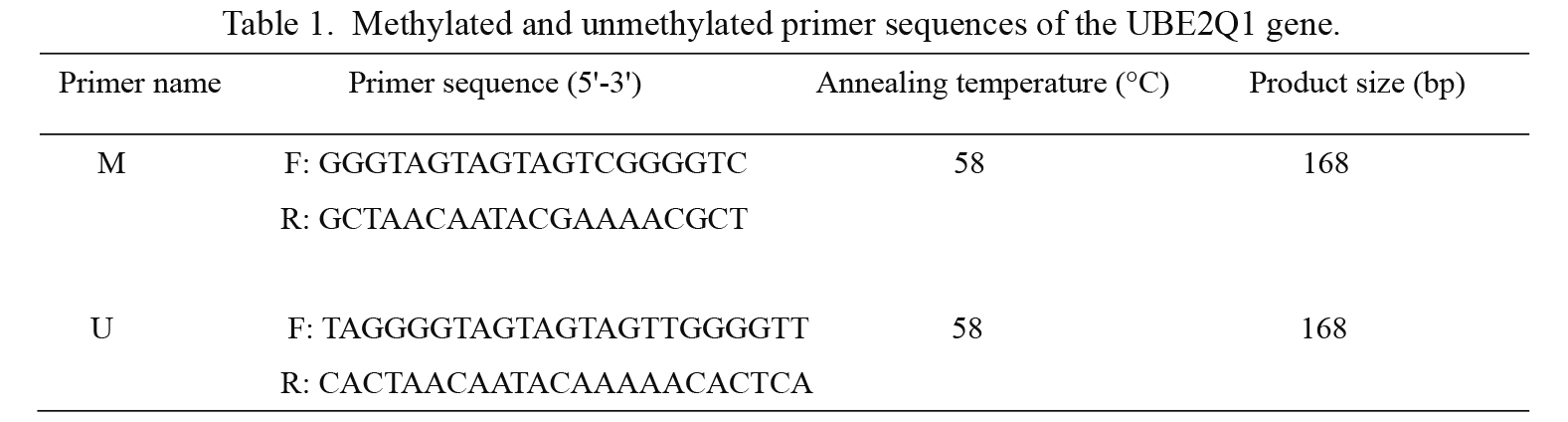
Sequencing primers for the methylated and unmethylated UBE2Q1 promoters were designed previously (Mokarram et al. 2015), and the primer sequences are shown in Table 1. The total MSP reaction mixture included 2 µl of modified DNA, 0.5 µl of the forward and reverse primers (10 µM), 12.5 µl of the ZymoTaqTM premix (Zymo Research Corp., CA, USA) and 9.5 µl of nuclease-free water to obtain a total volume of 25 µl. The PCR protocol was as follows: initial denaturation at 95°C for 5 min, followed by 45 cycles of denaturation at 95°C for 45 s, annealing at 58°C for 45 s, extension at 72°C for 60 s and a final extension at 72°C for 10 min. The PCR amplified products were electrophoresed on 2% agarose gels, stained with Gel Red (Biotium, CA, USA) and visualized under UV illumination. The MSP assay for each sample was repeated three times. Water without DNA was used as a negative control. Methylation positivity was defined by the presence of the PCR product only in the M lane or in both the M and U lanes, and methylation negativity was defined by the presence of the obtained PCR product only in the U lane.
The statistical analyses were performed with SPSS 19.0 (Chicago, IL, USA). The Chi-square test was used to compare categorical variables, and Spearman’s rank order correlation test was used to evaluate the correlations between different variables. The receiver operating characteristic (ROC) curve and area under the curve (AUC) were analyzed to evaluate the clinical value of methylation. A two-sided p < 0.05 was considered statistically significant.
Results
Baseline clinical characteristics
A total of 180 patients were enrolled and randomized (HCC Group, n = 80; LC Group, n = 40; CHB Group, n = 40; HCs Group, n = 20). The 80 patients with HBV infection-related HCC included 62 males and 18 females, and the median age of these patients was 57.00 years (inter-quartile range = 46.00-61.75 years). The baseline characteristics of the different groups, such as their ALT, AST, TBIL, ALB, and AFP levels, are summarized in Table 2.
Methylation status of the UBE2Q1 gene in the serum of patients with HBV-related HCC
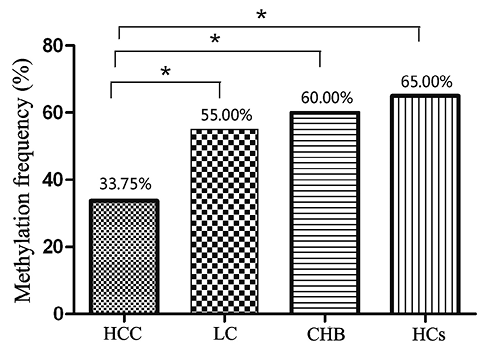
The methylation status of the UBE2Q1 gene in the serum of 180 patients was detected. Fig. 1 shows the typical representative gray value for methylation determination. Twenty-seven of the 80 (33.75%) HCC patients showed UBE2Q1 gene methylation, whereas, 22 of the 40 (55.00%) LC patients, 24 of the 40 (60.00%) CHB patients and 13 of the 20 (65.00%) HCs exhibited UBE2Q1 promoter methylation. The methylation frequency obtained for the HCC patients was significantly lower than found for the LC (χ2 = 4.98, p = 0.026) and CHB patients (χ2 = 7.52, p = 0.006) and HCs (χ2 = 6.51, p = 0.011). However, no significant difference in the methylation frequencies was found among the LC and CHB patients and HCs (χ2 = 0.58, p = 0.749; Fig. 2).
Correlation between the UBE2Q1 gene methylation status and clinicopathological parameters in HBV-related HCC
Eighty HBV-related HCC cases were analyzed to determine the associations between the DNA methylation of the UBE2Q1 gene and clinical features. The methylation level of the UBE2Q1 gene tended to be lower in patients with an advanced tumor node metastasis (TNM) stage (χ2 = 4.68, p = 0.030) and tumor size > 3 cm (χ2 = 5.09, p = 0.024; Table 3). The associations among methylation status, TNM stage, and tumor size were further analyzed by Spearman’s correlation coefficient. The results indicated a significant negative correlation between the methylation frequency and the TNM stage (rs = −0.30, p = 0.008), but a significant negative correlation was not found between the methylation frequency and tumor size (rs = −0.20, p = 0.080). No other significant association of the methylation frequency of the UBE2Q1 gene with clinicopathological parameters, such as gender, age, HBeAg, CTP class and venous invasion, was found (p > 0.05; Table 3).

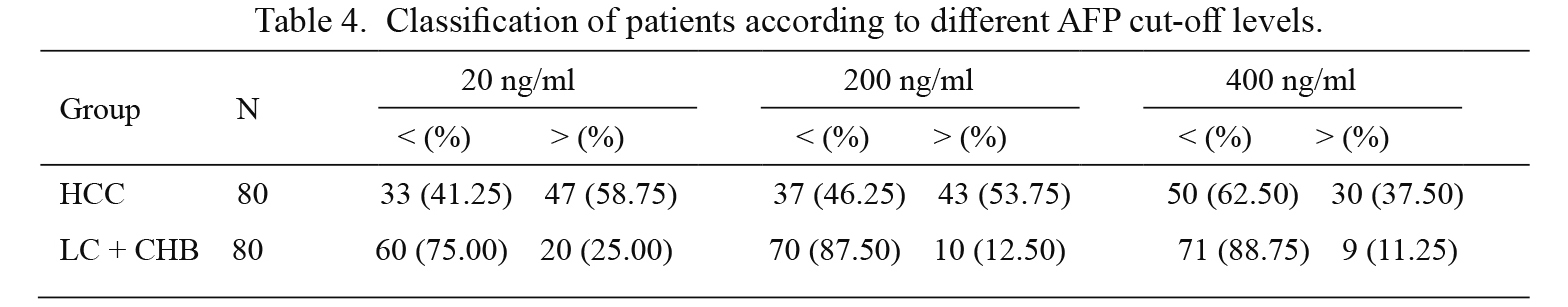
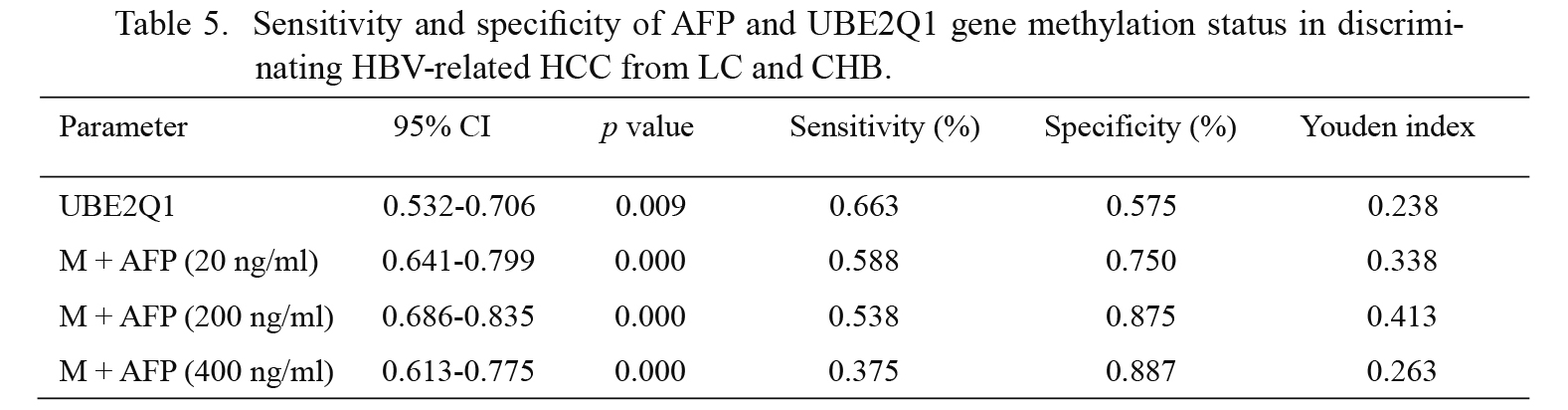
ROC curves were constructed to evaluate the performance of the UBE2Q1 gene methylation and serum AFP as biomarkers for distinguishing HCC from LC and CHB. An aberrant UBE2Q1 gene methylation status showed 66.3% sensitivity and 57.5% specificity in discriminating HCC from LC and CHB, and the diagnostic value of the serum AFP level showed sensitivity and specificity values of 53.8% and 87.5%, respectively. Furthermore, the diagnostic value of the UBE2Q1 gene methylation status combined with the AFP level evaluated using different cut-off points (20, 200 and 400 ng/ml) for the discrimination of HCC from LC and CHB was also calculated, and these analyses showed sensitivity and specificity values of 58.8% and 75.0%, 53.8% and 87.5%, and 37.5% and 88.7%, respectively (Tables 4 and 5).
The AUC values for UBE2Q1 gene methylation and the serum AFP level were 0.619 and 0.668, respectively (Fig. 3a). Moreover, a combinatorial analysis of the UBE2Q1 gene methylation status and the AFP level using different cut-off points specifically 20, 200 and 400 ng/ml, produced increased AUC values of 0.720, 0.760 and 0.694, respectively, in the discrimination of HCC from LC and CHB (Fig. 3b). These results indicate that the combination of the gene methylation status and AFP level might be optimal for the discrimination of HBV infection-related HCC from LC and CHB.
Discussion
In this study, we evaluated the methylation status of the UBE2Q1 gene in 80 HBV-related HCC patients, 40 LC patients, 40 CHB patients and 20 HCs. The results demonstrated that the methylation frequency of the UBE2Q1 gene was significantly lower in the sera of HBV-related HCC patients than in the sera of LC and CHB patients and HCs. However, no significant difference in methylation frequency was detected among the LC and CHB patients and HCs. In HBV-related HCC, aberrant methylation of the UBE2Q1 gene was significantly negatively correlated with the TNM stage. The combination of the serum UBE2Q1 gene methylation status with AFP using different cut-off points, specifically 20, 200 and 400 ng/ml, increased the AUC (0.720, 0.760 and 0.694, respectively), demonstrating that the combination of these biomarkers improves the discrimination of HCC from LC and CHB.
Epigenetic aberrations are strongly associated with the initiation and development of HCC (Khan et al. 2017). DNA methylation, which is the most extensively studied epigenetic modification, plays an important role in genome regulation (Bestor and Coxon 1993). DNA methylation modification can positively or negatively affect gene expression (Berger 2007). The exploration of epigenetic changes correlated with HCC is important for the design of better diagnostic and therapeutic methods. The ubiquitination enzymes include ubiquitin-activating enzyme (E1), ubiquitin-conjugating enzyme (E2) and ubiquitin ligases (E3). UBE2Q1, serving as E2, can transfer ubiquitin from E1 to E3 and other substrates and thereby regulate the degradation and function of the substrate protein (Grzmil et al. 2013).
Our study provides the first demonstration of aberrant hypomethylation of the UBE2Q1 gene in the serum of HBV-related HCC patients, and this methylation level was significantly lower than that found in LC and CHB patients. Hypomethylation of the UBE2Q1 promoter might negatively affect UBE2Q1 gene expression. Indeed, upregulated expression of the UBE2Q1 gene is associated with enhanced cell proliferation and poor prognosis in human HCC (Chang et al. 2015). Moreover, we found that the methylation frequency of the UBE2Q1 gene was significantly lower in HCC patients with a tumor size > 3 cm (χ2 = 5.09, p = 0.024) than in those with tumors ≤ 3 cm, and a negative correlation was found between UBE2Q1 gene methylation and the TNM stage, which is consistent with the results obtained in several other studies. Cheng et al. (2010) reported that DNA methylation is associated with tumor stage, and low levels of MT1M have been correlated with the clinical TNM grade (Mao et al. 2012). Qiu et al. (2016) also found a positive correlation between TSPYL5 methylation and advanced TNM stage. These results indicate that UBE2Q1 gene hypomethylation is closely associated with the progression of HBV-related HCC. Our findings reveal that methylation alterations of the UBE2Q1 gene might be used for disease detection and for the prediction of disease progression. Thus, UBE2Q1 might serve as a new biomarker for the surveillance of HBV-related HCC.
Chronic hepatitis B is closely related to the development of liver cirrhosis and HCC. As a result, CHB patients must be actively monitored and treated. HCC is one of the most serious malignancies worldwide because of a lack of early diagnosis and poor prognosis (Schafer and Sorrell 1999). The mean onset age of liver cirrhosis with chronic hepatitis B is 43.6 (24-69) years (Peng et al. 2012), and male Asian HBV patients older than 40 years and female Asian HBV patients older than 50 years are at high risk of developing HCC (Bruix and Sherman 2005). The age of the HCC and LC patients included in the present study was similar, and therefore, the identification of effective surveillance parameters for HCC is necessary. AFP is a relatively specific tumor marker for HCC, but measurement of the AFP concentration is not obligatory because this test is relatively insensitive when used for early detection (Malek et al. 2014). DNA methylation is an important early event in carcinogenesis, and the detection of methylated DNA has been recognized as a biomarker for the prediction of cancer initiation and progression. Although its detection lacks adequate sensitivity and specificity, the AFP level is clearly elevated in most HCC patients in areas of China and the Asia Pacific region and has thus been widely used as a serum biomarker for the screening of HCC (Bruix and Sherman 2005). The widely used, international cut-off values for AFP are 20, 200 and 400 ng/ml. In our study, the comparison of AFP alone with the combination of the UBE2Q1 gene methylation and the AFP level using different cut-off values increased the AUC. Therefore, as reported by Yang et al. (2014), the combination of the AFP level with aberrant DNA methylation enhance the diagnostic value of each individual biomarker for the discrimination of HBV-related HCC patients from LC and CHB patients.
The current study has several limitations. First, we only enrolled HBV-related HCC patients, and the limited number of these patients might have affected the accuracy of the conclusions. In China, some HCC patients develop HCC due to other pathogenic factors, such as HCV and alcoholism. Therefore, future studies should explore the methylation status of the UBE2Q1 gene in a larger sample size of patients who developed HCC due to different causes. Second, HBV-related HCC patients should be subjected to follow-up, which could provide long-term clinical profiles for analyzing the correlation between UBE2Q1 gene methylation and HCC prognosis. Third, the molecular mechanism for modulating UBE2Q1 promoter methylation requires further research.
In conclusion, our data show that hypomethylation of the UBE2Q1 gene is negatively associated with the TNM stage in HBV-related HCC. Moreover, the combination of UBE2Q1 gene methylation with the AFP level evaluated using cut-off points 20 and 200 ng/ml enhances the diagnostic value of these biomarkers for the discrimination of HBV-related HCC patients from LC and CHB patients. Thus, hypomethylation of the UBE2Q1 gene in serum could potentially be used as a biomarker in HCC.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (81371832), the Key Project of Chinese Ministry of Science and Technology (2012ZX10002007 and 2013ZX10002001), the Science and Technology Development Plan of Shandong Province (2014GSF118068), the Qingdao People’s Livelihood Science and Technology Project (15-9-2-91-NSH), and A Project of Shandong Province Higher Educational Science and Technology Program (J16LL03).
Conflict of Interest
The authors declare no conflict of interest.
References
-
Berger,
S.L.
(2007) The complex language of chromatin regulation during transcription. Nature, 447, 407-412.
-
Bestor,
T.H. &
Coxon,
A.
(1993) Cytosine methylation: the pros and cons of DNA methylation. Curr. Biol., 3, 384-386.
-
Bruix,
J. &
Sherman,
M.
(2005) Management of hepatocellular carcinoma. Hepatology, 42, 1208-1236.
-
Bruix,
J. &
Sherman,
M.
(2011) Management of hepatocellular carcinoma: an update. Hepatology, 53, 1020-1022.
-
Chan,
T.A. &
Baylin,
S.B.
(2012) Epigenetic biomarkers. Curr. Top. Microbiol. Immunol., 355, 189-216.
-
Chang,
R.,
Wei,
L.,
Lu,
Y.,
Cui,
X.,
Lu,
C.,
Liu,
L.,
Jiang,
D.,
Xiong,
Y.,
Wang,
G.,
Wan,
C. &
Qian,
H.
(2015) Upregulated expression of ubiquitin-conjugating enzyme E2Q1 (UBE2Q1) is associated with enhanced cell proliferation and poor prognosis in human hapatocellular carcinoma. J. Mol. Histol., 46, 45-56.
-
Chen,
D. &
Dou,
Q.P.
(2010) The ubiquitin-proteasome system as a prospective molecular target for cancer treatment and prevention. Curr. Protein Pept. Sci., 11, 459-470.
-
Cheng,
Y.,
Zhang,
C.,
Zhao,
J.,
Wang,
C.,
Xu,
Y.,
Han,
Z.,
Jiang,
G.,
Guo,
X.,
Li,
R.,
Bu,
X.,
Wu,
M. &
Wei,
L.
(2010) Correlation of CpG island methylator phenotype with poor prognosis in hepatocellular carcinoma. Exp. Mol. Pathol., 88, 112-117.
-
Chinese Society of Hepatology and Chinese Society of Infectious Diseases, Chinese Medical Association
(2011) The guideline of prevention and treatment for chronic hepatitis B (2010 version). Zhonghua Gan Zang Bing Za Zhi, 19, 13-24.
-
Cokus,
S.J.,
Feng,
S.,
Zhang,
X.,
Chen,
Z.,
Merriman,
B.,
Haudenschild,
C.D.,
Pradhan,
S.,
Nelson,
S.F.,
Pellegrini,
M. &
Jacobsen,
S.E.
(2008) Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature, 452, 215-219.
-
Ding,
F.,
Xiao,
H.,
Wang,
M.,
Xie,
X. &
Hu,
F.
(2014) The role of the ubiquitin-proteasome pathway in cancer development and treatment. Front. Biosci. (Landmark Ed.), 19, 886-895.
-
Dou,
C.Y.,
Fan,
Y.C.,
Cao,
C.J.,
Yang,
Y. &
Wang,
K.
(2016) Sera DNA methylation of CDH1, DNMT3b and ESR1 promoters as biomarker for the early diagnosis of hepatitis B virus-related hepatocellular carcinoma. Dig. Dis. Sci., 61, 1130-1138.
-
El-Serag,
H.B. &
Rudolph,
K.L.
(2007) Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology, 132, 2557-2576.
-
Forner,
A.,
Llovet,
J.M. &
Bruix,
J.
(2012) Hepatocellular carcinoma. Lancet, 379, 1245-1255.
-
Grzmil,
P.,
Altmann,
M.E.,
Adham,
I.M.,
Engel,
U.,
Jarry,
H.,
Schweyer,
S.,
Wolf,
S.,
Manz,
J. &
Engel,
W.
(2013) Embryo implantation failure and other reproductive defects in Ube2q1-deficient female mice. Reproduction, 145, 45-56.
-
Jahr,
S.,
Hentze,
H.,
Englisch,
S.,
Hardt,
D.,
Fackelmayer,
F.O.,
Hesch,
R.D. &
Knippers,
R.
(2001) DNA fragments in the blood plasma of cancer patients: quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res., 61, 1659-1665.
-
Khan,
F.S.,
Ali,
I.,
Afridi,
U.K.,
Ishtiaq,
M. &
Mehmood,
R.
(2017) Epigenetic mechanisms regulating the development of hepatocellular carcinoma and their promise for therapeutics. Hepatol. Int., 11, 45-53.
-
Kim,
J.S.,
Han,
J.,
Shim,
Y.M.,
Park,
J. &
Kim,
D.H.
(2005) Aberrant methylation of H-cadherin (CDH13) promoter is associated with tumor progression in primary nonsmall cell lung carcinoma. Cancer, 104, 1825-1833.
-
Lachenmayer,
A.,
Alsinet,
C.,
Chang,
C.Y. &
Llovet,
J.M.
(2010) Molecular approaches to treatment of hepatocellular carcinoma. Dig. Liver Dis., 42, S264-S272.
-
Laursen,
L.
(2014) A preventable cancer. Nature, 516, S2-S3.
-
Lecomte,
T.,
Ceze,
N.,
Dorval,
E. &
Laurent-Puig,
P.
(2010) Circulating free tumor DNA and colorectal cancer. Gastroenterol. Clin. Biol., 34, 662-681.
-
Malek,
N.P.,
Schmidt,
S.,
Huber,
P.,
Manns,
M.P. &
Greten,
T.F.
(2014) The diagnosis and treatment of hepatocellular carcinoma. Dtsch. Arztebl. Int., 111, 101-106.
-
Mao,
J.,
Yu,
H.,
Wang,
C.,
Sun,
L.,
Jiang,
W.,
Zhang,
P.,
Xiao,
Q.,
Han,
D.,
Saiyin,
H.,
Zhu,
J.,
Chen,
T.,
Roberts,
L.R.,
Huang,
H. &
Yu,
L.
(2012) Metallothionein MT1M is a tumor suppressor of human hepatocellular carcinomas. Carcinogenesis, 33, 2568-2577.
-
Mohamed,
N.A.,
Swify,
E.M.,
Amin,
N.F.,
Soliman,
M.M.,
Tag-Eldin,
L.M. &
Elsherbiny,
N.M.
(2012) Is serum level of methylated RASSF1A valuable in diagnosing hepatocellular carcinoma in patients with chronic viral hepatitis C? Arab J. Gastroenterol., 13, 111-115.
-
Mokarram,
P.,
Shakiba-Jam,
F.,
Kavousipour,
S.,
Sarabi,
M.M. &
Seghatoleslam,
A.
(2015) Promoter methylation status of two novel human genes, UBE2Q1 and UBE2Q2, in colorectal cancer: a new finding in Iranian patients. Asian Pac. J. Cancer Prev., 16, 8247-8252.
-
Nishida,
N. &
Goel,
A.
(2011) Genetic and epigenetic signatures in human hepatocellular carcinoma: a systematic review. Curr. Genomics, 12, 130-137.
-
Peng,
C.Y.,
Chien,
R.N. &
Liaw
Y.F.
(2012) Hepatitis B virus-related decompensated liver cirrhosis: benefits of antiviral therapy. J. Hepatol., 57, 442-450.
-
Perret,
C.
(2011) Methylation profile as a new tool for classification of hepatocellular carcinoma. J. Hepatol., 54, 602-603.
-
Qiu,
X.,
Hu,
B.,
Huang,
Y.,
Deng,
Y.,
Wang,
X. &
Zheng,
F.
(2016) Hypermethylation of ACP1, BMP4, and TSPYL5 in hepatocellular carcinoma and their potential clinical significance. Dig. Dis. Sci., 61, 149-157.
-
Schafer,
D.F. &
Sorrell,
M.F.
(1999) Hepatocellular carcinoma. Lancet, 353, 1253-1257.
-
Seghatoleslam,
A.,
Monabati,
A.,
Bozorg-Ghalati,
F.,
Nikseresht,
M.,
Bordbar,
M.R.,
Rahvar,
M. &
Owji,
A.A.
(2012) Expression of UBE2Q2, a putative member of the ubiquitin-conjugating enzyme family in pediatric acute lymphoblastic leukemia. Arch. Iran. Med., 15, 352-355.
-
Shafiee,
S.M.,
Rasti,
M.,
Seghatoleslam,
A.,
Azimi,
T. &
Owji,
A.A.
(2015) UBE2Q1 in a human breast carcinoma cell line: overexpression and interaction with p53. Asian Pac. J. Cancer Prev., 16, 3723-3727.
-
Shafiee,
S.M.,
Seghatoleslam,
A.,
Nikseresht,
M.,
Hosseini,
S.V.,
Alizadeh-Naeeni,
M.,
Safaei,
A. &
Owji,
A.A.
(2013) UBE2Q1 expression in human colorectal tumors and cell lines. Mol. Biol. Rep., 40, 7045-7051.
-
Sun,
F.K.,
Fan,
Y.C.,
Zhao,
J.,
Zhang,
F.,
Gao,
S.,
Zhao,
Z.H.,
Sun,
Q. &
Wang,
K.
(2013) Detection of TFPI2 methylation in the serum of hepatocellular carcinoma patients. Dig. Dis. Sci., 58, 1010-1015.
-
Um,
T.H.,
Kim,
H.,
Oh,
B.K.,
Kim,
M.S.,
Kim,
K.S.,
Jung,
G. &
Park,
Y.N.
(2011) Aberrant CpG island hypermethylation in dysplastic nodules and early HCC of hepatitis B virus-related human multistep hepatocarcinogenesis. J. Hepatol., 54, 939-947.
-
Venturelli,
S.,
Berger,
A.,
Weiland,
T.,
Essmann,
F.,
Waibel,
M.,
Nuebling,
T.,
Hacker,
S.,
Schenk,
M.,
Schulze-Osthoff,
K.,
Salih,
H.R.,
Fulda,
S.,
Sipos,
B.,
Johnstone,
R.W.,
Lauer,
U.M. &
Bitzer,
M.
(2013) Differential induction of apoptosis and senescence by the DNA methyltransferase inhibitors 5-azacytidine and 5-aza-2′-deoxycytidine in solid tumor cells. Mol. Cancer Ther., 12, 2226-2236.
-
Villanueva,
A.,
Minguez,
B.,
Forner,
A.,
Reig,
M. &
Llovet,
J.M.
(2010) Hepatocellular carcinoma: novel molecular approaches for diagnosis, prognosis, and therapy. Annu. Rev. Med., 61, 317-328.
-
Yang,
Y.,
Fan,
Y.C.,
Gao,
S.,
Dou,
C.Y.,
Zhang,
J.J.,
Sun,
F.K. &
Wang,
K.
(2014) Methylated cysteine dioxygenase-1 gene promoter in the serum is a potential biomarker for hepatitis B virus-related hepatocellular carcinoma. Tohoku J. Exp. Med., 232, 187-194.
-
Yates,
L.R. &
Campbell,
P.J.
(2012) Evolution of the cancer genome. Nat. Rev. Genet., 13, 795-806.
-
Zhong,
J.L. &
Huang,
C.Z.
(2016) Ubiquitin proteasome system research in gastrointestinal cancer. World J. Gastrointest. Oncol., 8, 198-206.