Abstract
Insufficient intake of polyunsaturated fatty acids (PUFA) causes fatty liver. The mechanism responsible is primarily related to increased lipogenesis and decreased FA degradation based on rodent studies. However, these studies were limited by the fact that the typical PUFA-deficient diets contained insufficient amounts of long-chain FA, the PUFA-containing diets were primarily composed of n-3 PUFA-enriched oil, and the intake of PUFA was excessive compared with the physiological requirement. To address these issues, mice were fed a PUFA-deficient diet containing long-chain FA at a standard fed level and then were orally fed a n-3/n-6-balanced PUFA-containing oil [PUFA (+)] or a PUFA-deficient oil [PUFA (−)] at physiological relevant levels (0.1 mL/mouse/2d). We compared these groups and examined whether fatty liver in PUFA deficiency was attributable to both the effects of increased lipogenesis and decreased FA catabolism. Compared with the PUFA (+) group, the PUFA (−) group showed increases in liver triglyceride and serum FA content. Hepatic gene expression of several mitochondrial β-oxidation enzymes, the serum 3-hydroxybutyrate level, and DNA-binding ability of peroxisome proliferator-activated receptor α (PPARα) were increased in the PUFA (+) group, whereas these adaptive responses were significantly attenuated in the PUFA (−) group. The hepatic expression of typical lipogenesis genes did not differ between the groups. Therefore, fatty liver in PUFA deficiency is attributable to suppression of the FA-degrading system probably from decreased PPARα adaptive responsiveness, and PUFA may be an essential factor for PPARα functioning. This finding is helpful for managing clinical situations having a risk of PUFA deficiency.
Introduction
Polyunsaturated fatty acids (PUFA) from the n-6 and n-3 series, including primarily linoleic acid (18:2, n-6) and α-linolenic acid (18:3, n-3), are essential nutrients for mammals (Spector and Kim 2015). These essential FA are the basic components of biological membranes and are important in maintaining cellular structure and function. PUFA are metabolized to the hormone-like chemicals eicosanoids, which regulate diverse biological functions including inflammation, immune responses, and reproduction (Funk 2001; Jump 2002). Insufficient intake of PUFA can lead to the development of deficiency symptoms, such as growth retardation, skin injury, reproductive impairment, and fatty liver (Burr and Burr 1929, 1930; Werner et al. 2005).
Several clinical studies have suggested that low intake of PUFA with high intake of saturated FA is related to the development of non-alcoholic fatty liver disease (NAFLD). The patients with NAFLD tend to prefer diets containing high saturated FA and low PUFA compared with the general population (Musso et al. 2003; Toshimitsu et al. 2007). Related to this dietary preference, the livers of NAFLD patients contain a lower ratio of certain PUFA species, such as arachidonic acid, eicosapentaenoic acid (EPA), and docosahexaenoic acid (DHA), in their total lipid extracts and triglyceride (TG) fractions compared with normal livers (Puri et al. 2007). Additionally, a recent study investigating NAFLD patients including those with steatohepatitis [i.e., NASH; fatty liver with inflammation and hepatocyte ballooning (Nagaya et al. 2010)] and simple steatosis [fatty liver without apparent histological features of NASH (Nagaya et al. 2010)] reported that the NASH patients’ livers contained a lower ratio of these PUFA species than those of the simple steatosis group, and that these PUFA profile differences were related to expression changes in several genes related to lipid metabolism (Arendt et al. 2015). These findings suggest that decreasing the levels of PUFA in diets and livers and the alteration of hepatic PUFA metabolism affect the disease progression of NAFLD. Therefore, understanding the physiological roles of PUFA in liver is important when considering new treatments toward NAFLD and related diseases.
The results from many rodent studies have reported the beneficial effects of PUFA in hepatic lipid metabolism. The most noticeable effect of PUFA is generally recognized as suppression of lipogenesis, which is primarily attributable to down-regulation of sterol regulatory element-binding protein-1c (SREBP-1c) (Kim et al. 1999; Mater et al. 1999; Sekiya et al. 2003; Tanaka et al. 2010), the master transcription factor for genes involved in FA and TG synthesis (Horton et al. 2002). PUFA are also known to stimulate FA degradation by activating peroxisome proliferator-activated receptor α (PPARα) (Ren et al. 1997; Martin et al. 2007), which is a ligand-dependent nuclear receptor that regulates the transcription of genes involved in FA β-oxidation (Aoyama et al. 1998; Kamijo et al. 2007). These two effects are proposed as the main mechanisms responsible for the liver TG-lowering action of PUFA.
A PUFA-deficient diet is known to induce fatty liver, in which the underlying mechanism is considered to be enhanced lipogenesis and decreased FA degradation (Alwayn et al. 2004; Werner et al. 2005). However, previous studies have some limitations in their experimental conditions. For example, to induce a PUFA-deficient condition, the previous studies generally used two types of PUFA-deficient diets: a fat-free diet or a 4-5% (w/w) hydrogenated coconut oil (HCO)-containing diet (Martin et al. 2007; Ducheix et al. 2013). However, these diets contain very small amounts of long-chain FA (< 1.5% in diet, w/w) compared with standard diets (4-7% in diet, w/w). Long-chain FA are necessary for maintaining cellular function and structure, and their insufficient intake affects several aspects of hepatic metabolism including lipogenesis (Shillabeer et al. 1990; Hudgins et al. 1996). Thus, the previously reported findings in PUFA-deficient diet-fed animals may be derived from the effects of both PUFA and long-chain FA deficiencies. Additionally, the previous studies showing the efficacy of PUFA used mainly fish oil as the PUFA-containing diet (Ren et al. 1997; Sekiya et al. 2003). Fish oil contains an abundant amount of n-3 PUFA, particularly EPA and DHA. Thus, the results may be biased by the specific effects brought about by the presence of n-3 PUFA. Third, the intake of PUFA-containing oil in the previous studies (> 4% in diet, w/w) was considerably higher than the physiological requirement (1-2% in diet, 2.5-3.5% energy in total intake) (Reeves 1997; FAO, Food and Agriculture Organization of the United Nations 2010), therefore, the results may differ from the physiological effects of PUFA. Based on these concerns, it remains unclear whether fatty liver in PUFA deficiency is caused by the effects of both enhanced lipogenesis and decreased FA catabolism.
In the present study, we aimed to clarify the mechanism of the development of fatty liver under a PUFA deficient condition. To solve the above-mentioned issues, we used a 14% (w/w) HCO-containing diet, which contained long-chain FA at the standard level (4% in diet, w/w). We fed mice with this PUFA-deficient diet for 2 or 5 weeks and orally treated them with a n-3/n-6-balanced PUFA-containing oil [PUFA (+)] or PUFA-deficient oil [PUFA (−)] at physiologically relevant levels (0.1 mL/mouse/2d), throughout the feeding period. We compared these mouse groups and examined whether fatty liver in PUFA deficiency was attributable to the effects of both increased lipogenesis and decreased FA catabolism.
Materials and Methods
Mice and treatment
This study was carried out in accordance with the Regulations for Animal Experimentation of Shinshu University. The animal protocol was approved by the Committee for Animal Experiments of Shinshu University (Approval Number 270044). Based on the national regulations and guidelines, all experimental procedures were reviewed by the Committee for Animal Experiments and finally approved by the president of Shinshu University.
Seven- to eight-week-old male C57BL/6 strain mice were purchased from Charles River Laboratories Japan (Yokohama, Japan) and maintained in a controlled environment (22-23°C; 12-h light/dark cycle) with tap water and a 5% (w/w) crude fat-containing standard rodent chow (MF; Oriental Yeast, Tokyo, Japan) ad libitum.
At 10 to 11 weeks of age, the mice (25-29 g of body weight) were divided into five groups with no difference in body weight average. One group continued being fed the standard chow (n = 3, Chow group), and diet for the other four groups (n = 5 in each) was switched to a 14% (w/w) hydrogenated coconut oil (HCO)-containing PUFA deficient diet (AIN-93G based; Oriental Yeast) for 2 or 5 weeks. The FA composition of this diet was as follows: C6:0, 0.2%; C8:0, 2.6%; C10:0, 6.4%; C12:0, 63.3%; C14:0, 15.7%; C16:0, 5.3%; and C18:0, 6.5% in total FA. Throughout the HCO feeding period, the mice were orally treated with PUFA-containing oil (soybean oil plus linseed oil, mixed at 1:1, v/v) [PUFA (+) group] or PUFA-deficient oil (coconut oil) [PUFA (−) group] via gastric tube at 0.1 mL/mouse every other day (typically in the evening). The dose of these supplemental oils was calculated as approximately 1.4-1.8 mg/g of body weight/day. This dosing level was selected based on a past study (Burr and Burr 1930); namely, in case of rats, oral administration of PUFA-containing oils, such as corn oil and raw linseed oil, at about 1 mg/g of body weight/day can prevent apparent symptoms of PUFA deficiency upon feeding a fat-free diet. All oils were obtained from Wako Pure Chemical Industries (Osaka, Japan). After purchase, soybean oil and linseed oil were immediately mixed, aliquoted, and stored at −25°C. Coconut oil was also aliquoted and stored in a similar manner. These oil aliquots were thawed at 37°C for 1-2 min, just before oral treatment. The day before sacrifice, the food was withdrawn from the cages, and the mice were fasted overnight, anesthetized with isoflurane (Wako) exposure, and euthanized by exsanguination from the vena cava inferior. The blood was coagulated at room temperature for 30 min, cooled on ice, and centrifuged at 1,000 × g for 10 min at 4°C for collection of serum. The liver was perfused via the left ventricle with 20 mL of cold physiological saline containing 1 mM EDTA (Wako), then removed, weighed, and immediately frozen on dry ice. Serum and liver were stored at −75°C until biochemical analysis.
Measurements of lipids and a ketone body
Serum TG and free FA (FFA) concentrations were determined using Triglyceride E-test and NEFA C-test kits (Wako), respectively. The serum 3-hydroxybutyrate concentration was measured using a β-hydroxybutyrate assay kit (BioVision, Milpitas, CA, USA).
Liver tissues (40-50 mg) were sonicated in 9 volumes of ice-cold 50 mM sodium phosphate buffer (pH 7.4), and 50 µL of the sonicate were mixed with 18 volumes of hexane:isopropanol (3:2, v/v) (Wako) (Hara and Radin 1978) as described previously (Li et al. 2007). The resulting supernatant containing total lipids (first extract) was collected to a new tube by decantation, and the remaining precipitate was washed with 300 µL of hexane:isopropanol:water (60:40:5, v/v) by brief vortexing. The resulting supernatant (second extract) was collected and combined with the first extract. This combined extract was then washed with aqueous sodium sulfate as previously described (Hara and Radin 1978), to remove nonlipid contaminants. The lipid extract was dried at 37°C in a gentle stream of nitrogen gas, dissolved in hexane:isopropanol (3:2, v/v) containing 1% (w/v) Triton X-100 (Wako), and dried again. After addition of water, the lipids were finally solubilized in 1% (w/v) Triton X-100/water (Carr et al. 1993). This lipid sample was used for measuring the contents of liver TG and FFA.
Thin-layer chromatography (TLC) of liver lipids
As mentioned above, 50 µL of the liver sonicate were mixed with 18 volumes of hexane:isopropanol (3:2, v/v), and the total lipid extract (first and second extract mixture) was collected in a vial and dried. The lipid was dissolved in chloroform:methanol (2:1, v/v) (Wako) at 10 µg liver equivalent/µL. This lipid sample (150 µg liver equivalent) was spotted on a TLC plate (Whatman LHP-K; GE Healthcare UK, Little Chalfont, Buckinghamshire, England), developed with hexane:ether:acetate (80:20:1, v/v/v), and detected with charring reagent (Hara and Taketomi 1990). Authentic standards (TG, FA, diacylglycerol, and monoacylglycerol; Wako) were also spotted on the same plate and analyzed. The position of the lipid bands was determined from the bands of authentic standards.
Gene expression analysis
Liver tissues (50-60 mg) were homogenized in 1 mL of TRIzol reagent (Thermo Fisher Scientific K.K., Yokohama, Japan). The homogenate was vigorously mixed with chloroform and centrifuged for phase separation according to the manufacturer’s instructions. The RNA-containing upper aqueous layer was collected, and total RNA was purified using a PureLink RNA mini kit and DNase (Thermo). The integrity of the total RNA extract was confirmed by spectrophotometry (Nanodrop 2000; Thermo) and gel electrophoresis. The liver total RNA (1 µg) was reverse-transcribed using a Takara PrimeScript RT reagent kit (Takara Bio, Kusatsu, Japan). The cDNA (4 ng RNA equivalent) was analyzed by real-time polymerase chain reaction (PCR) (StepOne Plus; Thermo) using SYBR green reagent (Takara SYBR Premix Ex Taq II; Takara). The primer sets were designed using Primer Express (Thermo), Primer 3, and Primer-BLAST systems (Table 1). The data analysis was carried out using the delta Ct method, and the expression data of target genes was normalized to that of the Gapdh gene (encoding glyceraldehyde-3-phosphate dehydrogenase) and each was shown as the fold change relative to that of the Chow group.

Approximately 300 mg of liver tissues were placed on ice, minced using scissors, and homogenized in 20 volumes of ice-cold buffer [0.25 M sucrose, 25 mM Tris chloride (pH 7.4), 25 mM potassium chloride, 5 mM magnesium chloride, 0.5% (w/v) Triton X-100, 1 mM dithiothreitol, and protease inhibitor cocktail (Thermo)] using a glass cylinder with a Teflon pestle (Aoyama et al. 1989, 1990). After removing the cell debris, the homogenate was centrifuged at 1,000 × g for 10 min at 2°C. The supernatant was removed, and the crude nuclear pellet was treated with Optiprep solution (Cosmo Bio, Tokyo, Japan). The resulting concentrated nucleus was treated with an extraction buffer [0.42 M sodium chloride, 20 mM Hepes (pH 7.9), 1.5 mM magnesium chloride, 0.2 mM EDTA, 25% (v/v) glycerol, 1 mM dithiothreitol, and protease inhibitor cocktail] for 1 h on ice with occasional vortexing, and the nuclear protein was extracted. The protein concentration of the liver nuclear extract was determined using a BCA protein assay kit (Thermo), and 5 µg of protein was subjected to the DNA-binding assays for SREBP-1, PPARα, and PPARγ using SREBP-1, PPARα, and PPARγ transcription factor assay kits (Cayman Chemicals, Ann Arbor, MI, USA), respectively. These assay kits are based on the enzyme-linked immunosorbent assay system and use a specific DNA binding sequence-immobilized microplate and specific antibody for each target transcription factor. This assay system is non-radioactive and sensitive and has been recently utilized as an alternative method to the radioactive electrophoretic mobility shift assay system (Harada et al. 2016). The results from each DNA-binding assay are shown as the fold change relative to that of the Chow group.
Data and statistical analyses
Results are expressed as the mean ± standard deviation. Statistical analysis was performed using two-way analysis of variance (SPSS Statistics 17.0; IBM Corporation, Armonk, NY, USA). A probability value of less than 0.05 was considered to be statistically significant.
Results
Effect on hepatic fat accumulation
Food intake was not different among the groups. Body, liver, and epididymal fat weights were increased by HCO feeding in a time-dependent manner, but did not significantly differ between the PUFA (+) and PUFA (−) groups at either time point (Table 2). TLC analysis of hepatic simple lipids demonstrated that the intensity of TG bands was increased by HCO feeding in a time-dependent manner and this increase was greater in the PUFA (−) group than in the PUFA (+) group (Fig. 1A). Quantitative analysis of liver TG showed similar results with a significant difference between the PUFA (−) and PUFA (+) mice at 5 weeks (Fig. 1B). There was no significant difference in the content of liver FFA or serum TG among the groups. The serum FFA level tended to be increased by HCO feeding in a time-dependent manner, and the level was higher in the PUFA (−) than in the PUFA (+) groups at 5 weeks (Fig. 1C). These results suggest that hepatic TG accumulation and serum FFA elevation were greater in the PUFA (−) mice than in the PUFA (+) mice.

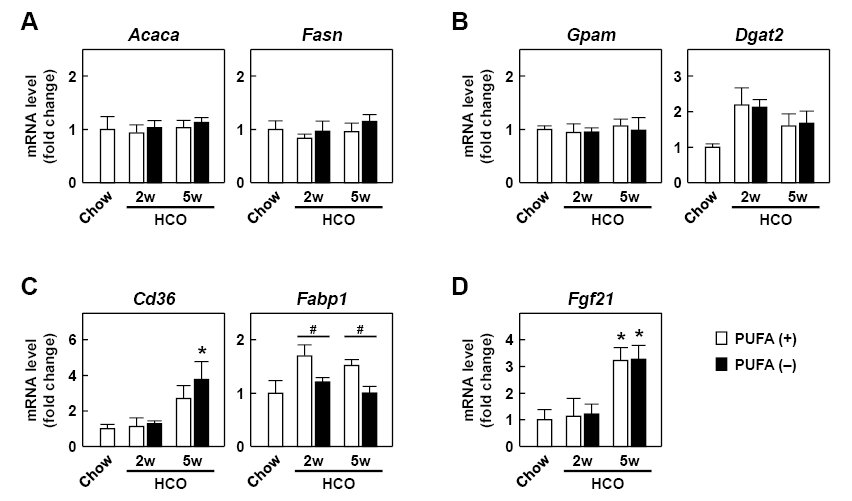
To understand the mechanism of the hepatic TG accumulation, we first examined hepatic expression of key genes involved in FA/TG synthesis. These included the Acaca and Fasn genes encoding acetyl-CoA carboxylase 1 and fatty acid synthase, respectively, which are the rate-limiting enzymes in de novo FA synthesis, and the Gpam and Dgat2 genes encoding glycerol-3-phosphate acyltransferase 1 and diacylglycerol acyltransferase 2, respectively, which are the key enzymes in the synthesis of TG from FA and glycerol. The expression levels of Acaca, Fasn, and Gpam were almost identical among the groups (Fig. 2A, B). The expression of Dgat2 was increased by HCO feeding, but did not differ between the PUFA (−) and PUFA (+) groups at either time point (Fig. 2B). These results suggest that alteration of lipogenesis gene expression scarcely influenced the hepatic TG accumulation in the PUFA (−) mice.
Effect on hepatic FA uptake and transport
Earlier studies reported that PUFA deficiency stimulated hepatic FA uptake from blood (Sinclair and Collins 1968; Fukazawa et al. 1970), thus, we examined this process. The level of the typical FA transporter gene Cd36, which encodes CD36 antigen/fatty acid translocase, was increased by HCO feeding in a time-dependent manner, especially in the PUFA (−) mice at 5 weeks. However, a significant difference between the PUFA (−) and PUFA (+) groups was not detected (Fig. 2C). The expression level of the Fabp1 gene encoding liver-type fatty acid-binding protein (L-FABP), a representative cellular FA transporter, was markedly higher in the PUFA (+) group, whereas this change was not detected in the PUFA (−) group and the level was the same as that in the Chow group (Fig. 2C). The expression levels of the Fgf21 gene encoding fibroblast growth factor-21, a hepatokine which stimulates lipolysis in white adipose tissue (Inagaki et al. 2007), were increased by HCO feeding especially at 5 weeks in the PUFA (−) and PUFA (+) groups, but these levels were not different between these groups (Fig. 2D).
Effect on hepatic FA catabolism
We next determined the gene expression of several mitochondrial FA β-oxidation enzymes (Fig. 3A) including long-chain acyl-CoA synthetase (Acsl1), carnitine palmitoyltransferase-I and -II (Cpt1a and 2), very-long-chain and long-chain acyl-CoA dehydrogenases (Acadvl and Acadl), and trifunctional protein α and β subunits (Hadha and Hadhb). Among these, the former three enzymes function in FA transport from the cytosol into the mitochondria and the remaining four enzymes catalyze the β-oxidation reaction. The expression level of the Acsl1 gene was markedly increased in the PUFA (+) group at both 2 and 5 weeks, whereas the level of the PUFA (−) group remained similar to that found for the Chow group. The expression level of the Cpt1a gene did not differ among the groups, whereas that of Cpt2 was significantly lower in the PUFA (−) mice at both time points compared with the Chow and PUFA (+) mice. The expression levels of Acadvl, Acadl, Hadha, and Hadhb were decreased in the PUFA (−) mice in a time-dependent manner, and a significant difference between the PUFA (−) and PUFA (+) groups was detected at the 5 weeks. The expression levels of Cpt2, Acadvl, Acadl, Hadha, and Hadhb were almost identical between the Chow and PUFA (+) groups. To further assess the effect of PUFA deficiency on hepatic mitochondrial β-oxidation, we measured serum ketone body levels, a typical indicator of the β-oxidation activity. Although the serum level of 3-hydroxybutyrate, the major ketone body in serum, was increased by HCO feeding, this increase was significantly suppressed in the PUFA (−) group compared with the PUFA (+) group (Fig. 3B).

Last, we examined SREBP-1c and PPARα. We also examined PPARγ, another PPAR subtype, since its expression and function can be induced in fatty liver (Gavrilova et al. 2003). The expression levels of the genes encoding SREBP-1c (Srebf1c) and PPARα (Ppara) tended to increase by HCO feeding, but were not significantly altered by either the feeding period or the existence of PUFA (Fig. 4A). The specific DNA-binding ability of nuclear SREBP-1c, which reflects its transcriptional activity, did not differ among the groups. In contrast, the specific DNA-binding ability of nuclear PPARα was increased in the PUFA (+) groups compared with the Chow group, whereas this increased transcriptional activity was not detected in the PUFA (−) group (Fig. 4B). This adaptive transcriptional response of PPARα appeared to correlate with the expression levels of the PPARα target gene (Figs. 2C and 3A; Fabp1, Acsl1, Cpt2, Acadvl, Acadl, Hadha, and Hadhb). By comparison, the expression of the PPARγ gene (Pparg) and its DNA-binding ability did not differ among the groups. These results suggest that the increased adaptive responsiveness of PPARα was suppressed in the PUFA (−) mice, which is associated with lower expression of several target genes involved in the β-oxidation system, followed by decrease of hepatic FA degradation. This condition might be the main cause of hepatic TG accumulation in the PUFA (−) mice.

Discussion
Past experimental conditions of PUFA deficiency were primarily based on insufficient intake of both PUFA and long-chain FA (Martin et al. 2007; Ducheix et al. 2013). Additionally, previous studies primarily used specific n-3 PUFA-enriched diets (Ren et al. 1997; Sekiya et al. 2003), in which the intake of PUFA was excessively higher than the physiological requirement (Reeves 1997; FAO 2010). Thus, the physiological mechanisms relevant to fatty liver in PUFA deficiency remain unclear.
To address these issues, we examined mice that received a long-chain FA-sufficient, PUFA-deficient diet in the presence or absence of PUFA supplementation at physiological required levels. We found that HCO feeding increased liver TG and serum FFA content time-dependently, indicating increased FA supply to the liver, and that these lipid changes were greater in the PUFA (−) mice than in the PUFA (+) mice. Compared with the Chow group, HCO feeding increased the PPARα DNA-binding ability, the expression of its targets Fabp1 and Acsl1, and the serum ketone body in the PUFA (+) mice, suggesting that PUFA (+) mice counteracted against oversupply of FA by enhancing their hepatic PPARα function. In contrast, the hepatic expression of six key genes in mitochondrial β-oxidation (Acsl1, Cpt2, Acadvl, Acadl, Hadha, and Hadhb), the level of serum 3-hydroxybutyrate, and the DNA-binding ability of PPARα were all significantly lower in the PUFA (−) group compared with the PUFA (+) group, suggesting that PUFA (−) mice could not sufficiently respond to this condition. Thus, PPARα-regulated signals were markedly influenced by the existence of PUFA, whereas the SREBP-1c-regulated lipogenesis signals (i.e., its DNA-binding ability and target gene expression involved in FA/TG synthesis) were minimally affected. Our overall findings are summarized in Fig. 5 and suggest that fatty liver in PUFA deficiency is primarily attributable to the suppression of the FA-degrading system induced by attenuation of PPARα adaptive responsiveness.
The poor adaptive responsiveness of PPARα, which was more prominent at 5 weeks than at 2 weeks in the PUFA (−) mice, may be attributed to the depletion of effective PPARα ligands derived from PUFA and their metabolites (Forman et al. 1997). Since the tissue content of PUFA is decreased along with the duration of PUFA-deficient treatment (Sinclair and Collins 1968), this time-dependent reduction of the intracellular content of PUFA-derived ligands might attenuate PPARα adaptive responsiveness. Another important point, the change in the transportability of PPARα ligands should be considered. The Fabp1 gene product L-FABP, a cytosolic protein, assists PPARα by transporting its ligands from the cytosol into the nucleus (Wolfrum et al. 2001), and L-FABP dysfunction suppresses the PPARα-regulated pathway, particularly under a high-fat diet feeding condition (Newberry et al. 2006). In the current study, the induction of the L-FABP gene was observed in the PUFA (+) mice at both 2 and 5 weeks, but not in the PUFA (−) groups at either time point. This poor induction of L-FABP might contribute to a lowering of the transportability of PPARα ligands resulting in attenuation of PPARα responsiveness.
The results from the current research showed that dietary PUFA is an essential factor for PPARα adaptive responsiveness. The PUFA-containing oil used in this study was a n-6/n-3 PUFA-balanced oil composed of equal amounts of linoleic and α-linolenic acids (about 30% (w/w), respectively), which are the most important PUFA in mammals (Spector and Kim 2015). Moreover, the dosage of this oil was much lower than that in past studies (Ren et al. 1997; Sekiya et al. 2003; Martin et al. 2007; Ducheix et al. 2013), and it was close to the physiological requirement (Reeves 1997; FAO 2010). As far as we know, there is no report showing that these two essential PUFA affect hepatic PPARα function in vivo at physiological intake levels. Therefore, the present findings reveal the physiological effect of dietary PUFA more accurately than the results from past studies.
Induction of lipogenesis gene expression was not apparent in the livers of PUFA (−) mice, a result that is different from previous results (Martin et al. 2007; Ducheix et al. 2013). This difference might be attributed to the difference in the content of long-chain FA in the diet. Several studies in humans and animals have demonstrated that dietary long-chain FA content is an important factor influencing hepatic lipogenesis (Shillabeer et al. 1990; Hudgins et al. 1996). Similar to our results, it is reported that rats fed a PUFA-deficient diet, which contained a standard fed level of long-chain FA, exhibited poor induction of hepatic FA synthesis activity irrespective of the development of marked fatty liver (Sinclair and Collins 1968). In contrast, rats fed a fat-free diet showed approximately five times higher hepatic FA synthesis activity (Sinclair and Collins 1968). These findings suggest that insufficient intake of long-chain FA might promote hepatic lipogenesis in the PUFA-deficient animals previously receiving a fat-free or 4-5% HCO-based diet (Martin et al. 2007; Ducheix et al. 2013). Furthermore, these past studies compared PUFA-deficient animals to PUFA-supplemented animals, which were administered strong anti-lipogenesis PUFA, EPA and DHA. These previously used experimental conditions may highlight the lipogenesis factors in the development process of fatty liver in PUFA-deficient animals.
Several studies have reported that hepatic FA uptake is enhanced by PUFA deficiency (Sinclair and Collins 1968; Fukazawa et al. 1970). Consistent with these reports, hepatic Cd36 expression levels were markedly increased in the PUFA (−) mice at 5 weeks. However, a significant difference in the Cd36 level was not detected between the PUFA (+) and PUFA (−) mice. Moreover, the change of another related FA uptake factor, Fabp1 expression, was opposite to that of Cd36 where a higher level was observed in the PUFA (+) mice and a lower level was observed in the PUFA (−) mice. Thus, we could not determine the effect of PUFA deficiency on hepatic FA uptake. Since the alteration of FA uptake is closely related to the development of fatty liver (Tailleux et al. 2012), it is necessary to clarify the detailed effects of dietary PUFA in this process.
The limitation of this study includes the use of a 14% HCO-containing diet, which is a high fat diet. HCO contains large amounts of medium-chain FA (> 70% in total FA). Since medium-chain FA are quickly metabolized in cells compared with long-chain FA (Schönfeld and Wojtczak 2016), the effect of this HCO-based high-fat diet on hepatic and whole-body fat accumulation is probably less than that of typical high-fat diets composed of long-chain FA-based oils. However, we cannot deny the possibility that feeding of HCO-based high-fat diets affects hepatic expression of several genes involved in lipid metabolism (Lin et al. 2005). Further studies using PUFA-deficient diets with a normal fat level, which consist of long-chain FA at the standard level, are necessary to confirm the results in the present study.
In conclusion, the present study demonstrated that fatty liver in PUFA deficiency is primarily associated with a decrease of PPARα adaptive responsiveness and the resulting attenuation of FA β-oxidation ability, but is minimally affected by induction of SREBP-1c-regulated lipogenesis action. The results from the current study also revealed that dietary PUFA is an essential factor for PPARα adaptive responsiveness in the liver. These findings may be helpful for considering new treatments against NAFLD and FA metabolism disorders and for managing other clinical situations where there is a risk of PUFA deficiency.
Acknowledgments
This work was supported in part by JSPS KAKENHI Grant Number 15K08264.
Conflict of Interest
The authors declare no conflict of interest.
References
-
Alwayn,
I.P.,
Javid,
P.J.,
Gura,
K.M.,
Nosé,
V.,
Ollero,
M. &
Puder,
M.
(2004) Do polyunsaturated fatty acids ameliorate hepatic steatosis in obese mice by SREPB-1 suppression or by correcting essential fatty acid deficiency. Hepatology, 39, 1176-1177.
-
Aoyama,
T.,
Hardwick,
J.P.,
Imaoka,
S.,
Funae,
Y.,
Gelboin,
H.V. &
Gonzalez,
F.J.
(1990) Clofibrate-inducible rat hepatic P450s IVA1 and IVA3 catalyze the ω- and (ω-1)-hydroxylation of fatty acids and the ω-hydroxylation of prostaglandins E1 and F2α. J. Lipid Res., 31, 1477-1482.
-
Aoyama,
T.,
Peters,
J.M.,
Iritani,
N.,
Nakajima,
T.,
Furihata,
K.,
Hashimoto,
T. &
Gonzalez,
F.J.
(1998) Altered constitutive expression of fatty acid-metabolizing enzymes in mice lacking the peroxisome proliferator-activated receptor α (PPARα). J. Biol. Chem., 273, 5678-5684.
-
Aoyama,
T.,
Yamano,
S.,
Waxman,
D.J.,
Lapenson,
D.P.,
Meyer,
U.A.,
Fischer,
V.,
Tyndale,
R.,
Inaba,
T.,
Kalow,
W. &
Gelboin,
H.V.
(1989) Cytochrome P-450 hPCN3, a novel cytochrome P-450 IIIA gene product that is differentially expressed in adult human liver. cDNA and deduced amino acid sequence and distinct specificities of cDNA-expressed hPCN1 and hPCN3 for the metabolism of steroid hormones and cyclosporine. J. Biol. Chem., 264, 10388-10395.
-
Arendt,
B.M.,
Comelli,
E.M.,
Ma,
D.W.,
Lou,
W.,
Teterina,
A.,
Kim,
T.,
Fung,
S.K.,
Wong,
D.K.,
McGilvray,
I.,
Fischer,
S.E. &
Allard,
J.P.
(2015) Altered hepatic gene expression in nonalcoholic fatty liver disease is associated with lower hepatic n-3 and n-6 polyunsaturated fatty acids. Hepatology, 61, 1565-1578.
-
Burr,
G.O. &
Burr,
M.M.
(1929) A new deficiency disease produced by the rigid exclusion of fat from the diet. J. Biol. Chem., 82, 345-367.
-
Burr,
G.O. &
Burr,
M.M.
(1930) On the nature and role of the fatty acids essential in nutrition. J. Biol. Chem., 86, 587-621.
-
Carr,
T.P.,
Andresen,
C.J. &
Rudel,
L.L.
(1993) Enzymatic determination of triglyceride, free cholesterol, and total cholesterol in tissue lipid extracts. Clin. Biochem., 26, 39-42.
-
Ducheix,
S.,
Montagner,
A.,
Polizzi,
A.,
Lasserre,
F.,
Marmugi,
A.,
Bertrand-Michel,
J.,
Podechard,
N.,
Al Saati,
T.,
Chétiveaux,
M.,
Baron,
S.,
Boué,
J.,
Dietrich,
G.,
Mselli-Lakhal,
L.,
Costet,
P.,
Lobaccaro,
J.M., et al.
(2013) Essential fatty acids deficiency promotes lipogenic gene expression and hepatic steatosis through the liver X receptor. J. Hepatol., 58, 984-992.
-
Food and Agriculture Organization of the United Nations (FAO)
(2010) Fats and fatty acids in human nutrition. Report of an expert consultation. FAO Food Nutr. Pap., 91, 1-166.
-
Forman,
B.M.,
Chen,
J. &
Evans,
R.M.
(1997) Hypolipidemic drugs, polyunsaturated fatty acids, and eicosanoids are ligands for peroxisome proliferator-activated receptors α and δ. Proc. Natl. Acad. Sci. USA, 94, 4312-4317.
-
Fukazawa,
T.,
Privett,
O.S. &
Takahashi,
Y.
(1970) Effect of essential fatty acid deficiency on release of triglycerides by the perfused rat liver. J. Lipid Res., 11, 522-527.
-
Funk,
C.D.
(2001) Prostaglandins and leukotrienes: advances in eicosanoid biology. Science, 294, 1871-1875.
-
Gavrilova,
O.,
Haluzik,
M.,
Matsusue,
K.,
Cutson,
J.J.,
Johnson,
L.,
Dietz,
K.R.,
Nicol,
C.J.,
Vinson,
C.,
Gonzalez,
F.J. &
Reitman,
M.L.
(2003) Liver peroxisome proliferator-activated receptor γ contributes to hepatic steatosis, triglyceride clearance, and regulation of body fat mass. J. Biol. Chem., 278, 34268-34276.
-
Hara,
A. &
Radin,
N.S.
(1978) Lipid extraction of tissues with a low-toxicity solvent. Anal. Biochem., 90, 420-426.
-
Hara,
A. &
Taketomi,
T.
(1990) Characterization and change of phospholipids in the aorta of Watanabe hereditable hyperlipidemic rabbit. Jpn. J. Exp. Med. 60, 311-318.
-
Harada,
M.,
Kamijo,
Y.,
Nakajima,
T.,
Hashimoto,
K.,
Yamada,
Y.,
Shimojo,
H.,
Gonzalez,
F.J. &
Aoyama,
T.
(2016) Peroxisome proliferator-activated receptor α-dependent renoprotection of murine kidney by irbesartan. Clin. Sci., 130, 1969-1981.
-
Horton,
J.D.,
Goldstein,
J.L. &
Brown,
M.S.
(2002) SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Invest., 109, 1125-1131.
-
Hudgins,
L.C.,
Hellerstein,
M.,
Seidman,
C.,
Neese,
R.,
Diakun,
J. &
Hirsch,
J.
(1996) Human fatty acid synthesis is stimulated by a eucaloric low fat, high carbohydrate diet. J. Clin. Invest., 97, 2081-2091.
-
Inagaki,
T.,
Dutchak,
P.,
Zhao,
G.,
Ding,
X.,
Gautron,
L.,
Parameswara,
V.,
Li,
Y.,
Goetz,
R.,
Mohammadi,
M.,
Esser,
V.,
Elmquist,
J.K.,
Gerard,
R.D.,
Burgess,
S.C.,
Hammer,
R.E.,
Mangelsdorf,
D.J. &
Kliewer,
S.A.
(2007) Endocrine regulation of the fasting response by PPARα-mediated induction of fibroblast growth factor 21. Cell Metab., 5, 415-425.
-
Jump,
D.B.
(2002) The biochemistry of n-3 polyunsaturated fatty acids. J. Biol. Chem., 277, 8755-8758.
-
Kamijo,
Y.,
Hora,
K.,
Kono,
K.,
Takahashi,
K.,
Higuchi,
M.,
Ehara,
T.,
Kiyosawa,
K.,
Shigematsu,
H.,
Gonzalez,
F.J. &
Aoyama,
T.
(2007) PPARα protects proximal tubular cells from acute fatty acid toxicity. J. Am. Soc. Nephrol., 18, 3089-3100.
-
Kim,
H.J.,
Takahashi,
M. &
Ezaki,
O.
(1999) Fish oil feeding decreases mature sterol regulatory element-binding protein 1 (SREBP-1) by down-regulation of SREBP-1c mRNA in mouse liver. A possible mechanism for down-regulation of lipogenic enzyme mRNAs. J. Biol. Chem., 274, 25892-25898.
-
Li,
G.,
Hu,
R.,
Kamijo,
Y.,
Nakajima,
T.,
Aoyama,
T.,
Inoue,
T.,
Node,
K.,
Kannagi,
R.,
Kyogashima,
M. &
Hara,
A.
(2007) Establishment of a quantitative, qualitative, and high-throughput analysis of sulfatides from small amounts of sera by matrix-assisted laser desorption ionization-time of flight mass spectrometry. Anal. Biochem., 362, 1-7.
-
Lin,
J.,
Yang,
R.,
Tarr,
P.T.,
Wu,
P.H.,
Handschin,
C.,
Li,
S.,
Yang,
W.,
Pei,
L.,
Uldry,
M.,
Tontonoz,
P.,
Newgard,
C.B. &
Spiegelman,
B.M.
(2005) Hyperlipidemic effects of dietary saturated fats mediated through PGC-1β coactivation of SREBP. Cell, 120, 261-273.
-
Martin,
P.G.,
Guillou,
H.,
Lasserre,
F.,
Déjean,
S.,
Lan,
A.,
Pascussi,
J.M.,
Sancristobal,
M.,
Legrand,
P.,
Besse,
P. &
Pineau,
T.
(2007) Novel aspects of PPARα-mediated regulation of lipid and xenobiotic metabolism revealed through a nutrigenomic study. Hepatology, 45, 767-777.
-
Mater,
M.K.,
Thelen,
A.P.,
Pan,
D.A. &
Jump,
D.B.
(1999) Sterol response element-binding protein 1c (SREBP1c) is involved in the polyunsaturated fatty acid suppression of hepatic S14 gene transcription. J. Biol. Chem., 274, 32725-32732.
-
Musso,
G.,
Gambino,
R.,
De Michieli,
F.,
Cassader,
M.,
Rizzetto,
M.,
Durazzo,
M.,
Fagà,
E.,
Silli,
B. &
Pagano,
G.
(2003) Dietary habits and their relations to insulin resistance and postprandial lipemia in nonalcoholic steatohepatitis. Hepatology, 37, 909-916.
-
Nagaya,
T.,
Tanaka,
N.,
Suzuki,
T.,
Sano,
K.,
Horiuchi,
A.,
Komatsu,
M.,
Nakajima,
T.,
Nishizawa,
T.,
Joshita,
S.,
Umemura,
T.,
Ichijo,
T.,
Matsumoto,
A.,
Yoshizawa,
K.,
Nakayama,
J.,
Tanaka,
E. &
Aoyama,
T.
(2010) Down-regulation of SREBP-1c is associated with the development of burned-out NASH. J. Hepatol., 53, 724-731.
-
Newberry,
E.P.,
Xie,
Y.,
Kennedy,
S.M.,
Luo,
J. &
Davidson,
N.O.
(2006) Protection against Western diet-induced obesity and hepatic steatosis in liver fatty acid-binding protein knockout mice. Hepatology, 44, 1191-1205.
-
Puri,
P.,
Baillie,
R.A.,
Wiest,
M.M.,
Mirshahi,
F.,
Choudhury,
J.,
Cheung,
O.,
Sargeant,
C.,
Contos,
M.J. &
Sanyal,
A.J.
(2007) A lipidomic analysis of nonalcoholic fatty liver disease. Hepatology, 46, 1081-1090.
-
Reeves,
P.G.
(1997) Components of the AIN-93 diets as improvements in the AIN-76A diet. J. Nutr., 127, 838S-841S.
-
Ren,
B.,
Thelen,
A.P.,
Peters,
J.M.,
Gonzalez,
F.J. &
Jump,
D.B.
(1997) Polyunsaturated fatty acid suppression of hepatic fatty acid synthase and S14 gene expression does not require peroxisome proliferator-activated receptor α. J. Biol. Chem., 272, 26827-26832.
-
Schönfeld,
P. &
Wojtczak,
L.
(2016) Short- and medium-chain fatty acids in energy metabolism: the cellular perspective. J. Lipid Res., 57, 943-954.
-
Sekiya,
M.,
Yahagi,
N.,
Matsuzaka,
T.,
Najima,
Y.,
Nakakuki,
M.,
Nagai,
R.,
Ishibashi,
S.,
Osuga,
J.,
Yamada,
N. &
Shimano,
H.
(2003) Polyunsaturated fatty acids ameliorate hepatic steatosis in obese mice by SREBP-1 suppression. Hepatology, 38, 1529-1539.
-
Shillabeer,
G.,
Hornford,
J.,
Forden,
J.M.,
Wong,
N.C. &
Lau,
D.C.
(1990) Hepatic and adipose tissue lipogenic enzyme mRNA levels are suppressed by high fat diets in the rat. J. Lipid Res., 31, 623-631.
-
Shimomura,
I.,
Shimano,
H.,
Horton,
J.D.,
Goldstein,
J.L. &
Brown,
M.S.
(1997) Differential expression of exons 1a and 1c in mRNAs for sterol regulatory element binding protein-1 in human and mouse organs and cultured cells. J. Clin. Invest., 99, 838-845.
-
Sinclair,
A.J. &
Collins,
F.D.
(1968) Fatty livers in rats deficient in essential fatty acids. Biochem. Biophys. Acta, 152, 498-510.
-
Spector,
A.A. &
Kim,
H.Y.
(2015) Discovery of essential fatty acids. J. Lipid Res., 56, 11-21.
-
Tailleux,
A.,
Wouters,
K. &
Staels,
B.
(2012) Roles of PPARs in NAFLD: potential therapeutic targets. Biochim. Biophys. Acta, 1821, 809-818.
-
Tanaka,
N.,
Zhang,
X.,
Sugiyama,
E.,
Kono,
H.,
Horiuchi,
A.,
Nakajima,
T.,
Kanbe,
H.,
Tanaka,
E.,
Gonzalez,
F.J. &
Aoyama,
T.
(2010) Eicosapentaenoic acid improves hepatic steatosis independent of PPARα activation through inhibition of SREBP-1 maturation in mice. Biochem. Pharmacol., 80, 1601-1612.
-
Toshimitsu,
K.,
Matsuura,
B.,
Ohkubo,
I.,
Niiya,
T.,
Furukawa,
S.,
Hiasa,
Y.,
Kawamura,
M.,
Ebihara,
K. &
Onji,
M.
(2007) Dietary habits and nutrient intake in non-alcoholic steatohepatitis. Nutrition, 23, 46-52.
-
Werner,
A.,
Havinga,
R.,
Bos,
T.,
Bloks,
V.W.,
Kuipers,
F. &
Verkade,
H.J.
(2005) Essential fatty acid deficiency in mice is associated with hepatic steatosis and secretion of large VLDL particles. Am. J. Physiol. Gastrointest. Liver Physiol., 288, G1150-G1158.
-
Wolfrum,
C.,
Borrmann,
C.M.,
Borchers,
T. &
Spener,
F.
(2001) Fatty acids and hypolipidemic drugs regulate peroxisome proliferator-activated receptors α- and γ-mediated gene expression via liver fatty acid binding protein: a signaling path to the nucleus. Proc. Natl. Acad. Sci. USA, 98, 2323-2328.