Abstract
Pulmonary alveolar proteinosis (PAP) is characterized by the accumulation of periodic acid-schiff stain-positive lipoproteinaceous materials in the alveolar space due to impaired surfactant clearance by alveolar macrophage. Autoimmune PAP is the most common form of PAP, but rarely accompanies collagen disease or sarcoidosis. We report here a rare case of autoimmune PAP preceded by systemic sclerosis and sarcoidosis. A 64-year-old woman was admitted to our hospital for blurred vision, muscle weakness of extremities, Raynaud’s phenomenon, and exertional dyspnea. We diagnosed her as having systemic sclerosis complicated with sarcoidosis. Chest computed tomography (CT) and transbronchial lung biopsy showed the findings of pulmonary fibrosis without PAP. We treated her with corticosteroid and intravenous cyclophosphamide therapy, followed by tacrolimus therapy. Thereafter, her symptoms improved except for exertional dyspnea, and she began to complain of productive cough thirteen months after corticosteroid and immunosuppressant therapy. On the second admission, a chest CT scan detected the emergence of crazy-paving pattern in bilateral upper lobes. Bronchoalveolar lavage (BAL) fluid with milky appearance and a lung biopsy specimen revealed acellular periodic acid-schiff stain-positive bodies. The serum titer of anti-granulocyte macrophage colony stimulating factor (GM-CSF) antibodies was elevated on first admission and remained high on second admission. We thus diagnosed her as having autoimmune PAP. Reducing the dose of immunosuppressive agents and repeating the segmental BAL resulted in the improvement of her symptoms and radiological findings. Immunosuppressant therapy may trigger the onset of autoimmune PAP in a subset of patients with systemic sclerosis and/or sarcoidosis.
Introduction
Pulmonary alveolar proteinosis (PAP) is a rare disease that is characterized by the accumulation of periodic acid-schiff (PAS) stain-positive lipoproteinaceous materials in the alveolar space due to impaired surfactant clearance by alveolar macrophage (Trapnell et al. 2003). PAP occurs in three clinically distinct forms: congenital, autoimmune, and secondary PAP. Autoimmune PAP is the most common form of PAP, which consists of approximately 90% of PAP (Trapnell et al. 2003). The anti-granulocyte macrophage colony stimulating factor (GM-CSF) antibodies are specifically detected in serum and bronchoalveolar lavage (BAL) from patients with autoimmune PAP (Kitamura et al. 1999, 2000). The autoantibodies neutralize the biological activity of GM-CSF and impair the clearance of pulmonary surfactant by macrophages, leading to the accumulation of surfactant proteins and cellular debris in the alveolar space and disruption of gas exchange (Kitamura et al. 1999; Uchida et al. 2004). Autoimmune PAP is a rare disease with the morbidity of 0.5 per 100,000 (Inoue et al. 2006). The prevalence rate of autoimmune disease in autoimmune PAP is reported to be 1.4-1.6% in the large cohort study (Seymour and Presneill 2002; Inoue et al. 2008). Details were reported in three cases of autoimmune PAP associated with collagen disease (Nagasawa et al. 2013; Kinehara et al. 2014; Imura et al. 2016) in the literature. There is only one case report of autoimmune PAP associated with sarcoidosis (Boerner et al. 2016). Herein, we reported a rare case of autoimmune PAP associated with systemic sclerosis and sarcoidosis. The serum level of anti-GM-CSF antibodies had been elevated two years before the onset of autoimmune PAP. Immunosuppressant therapy would trigger the development of autoimmune PAP in a patient with co-existed systemic sclerosis and sarcoidosis. The reduction of immunosuppressant therapy and repeated segmental BAL resulted in the remission.
Case Report
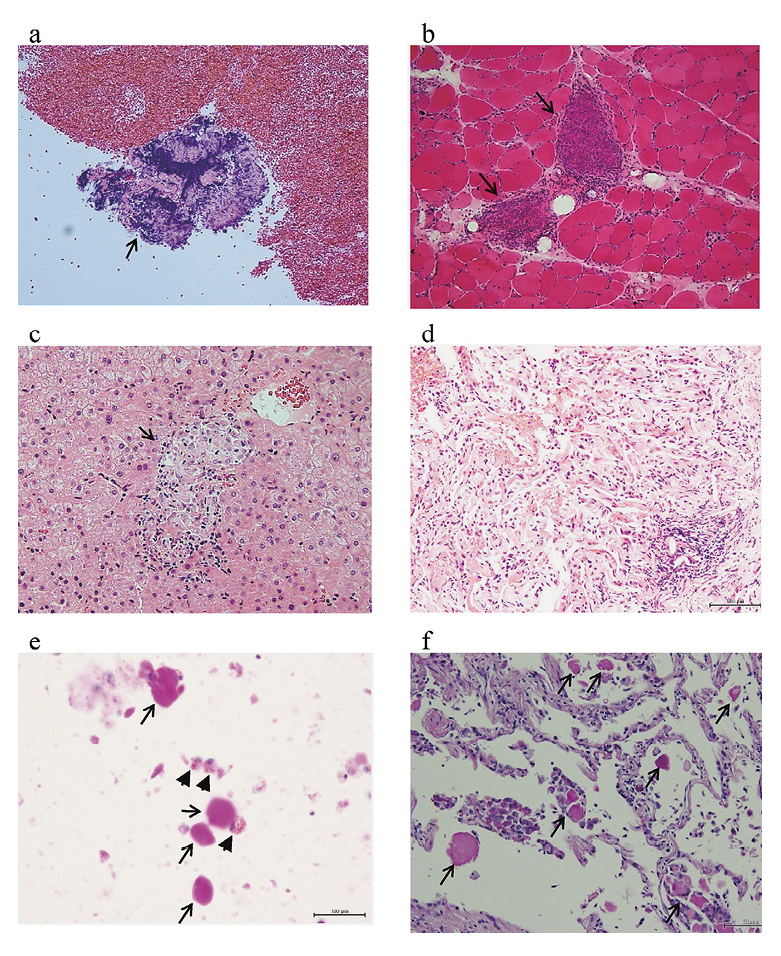
In January 2014, a 64-year-old woman was admitted to our hospital for blurred vision, muscle weakness of extremities, Raynaud’s phenomenon, and exertional dyspnea continuing since December 2012. She had a past medical history of reflux esophagitis. Her temperature was 36.7°C, pulse rate 84 beats/min, respiratory rate 22 breaths/min, and SpO2 96% at room air. An ophthalmologist diagnosed her as having bilateral uveitis which was suggestive of ocular sarcoidosis. We could observe sclerodermatous changes on facial and hand skin, and telangiectasia on buccal region of face and finger pads. Manual muscle testing (MMT) of proximal muscles of bilateral extremities was decreased to 4/5 in bilateral deltoid muscles and in both quadriceps muscles, and to 3/5 in both adductors muscles. Fine crackles were also heard in bilateral lower fields. The serum level of aspartate aminotransferase (AST), alanine aminotransferase (ALT), creatine phosphokinase (CPK), and aldolase were elevated to 70 IU/L, 33 IU/L, 763 IU/L, and 10.3 IU/L, respectively. The serum level of angiotensin converting enzyme (ACE) was within normal range (17 IU/mL/37℃). The serum lysozyme and soluble interleukin-2 receptor (sIL-2R) were elevated to 19.5 µg/mL and 1,233 IU/L, respectively. The serum levels of antinuclear antibodies (ANA) and anti-RNA polymerase III antibodies (ARA) were elevated to × 640 and × 714, respectively. The serum level of CEA was within normal range (1.4 ng/mL). The serum level of KL-6 was elevated to 7,353 U/mL (cut off value < 500 U/mL, Fig. 1). Bronchoalveolar lavage (BAL) against the right B5b revealed increased percentage of lymphocytes (41.5%) and CD4/CD8 ratio of 7.2. The appearance of bronchoalveolar lavage fluid (BALF) was normal. An arterial blood gas analysis showed a normal value of PaO2 (81 torr at room air). Pulmonary functional test (PFT) showed a decrease of %DLco to 49.6%. Within normal range were %VC and %FEV1: 88.2% and 79.5%, respectively. A computed tomography (CT) scan showed esophageal dilation, bilateral hilar and mediastinal lymphadenopathy, and reticular opacity in the bilateral lower lobes (Fig. 2a-c). Gallium-67 (67Ga) scintigraphy detected abnormal 67Ga accumulation in bilateral hilar and mediastinal lymph nodes, liver, and bilateral upper arms (Fig. 3). Biopsy specimens of mediastinal lymph nodes, the muscle of the left upper arm, and the liver showed non-caseating epithelioid cell granulomas consistent with sarcoidosis (Fig. 4a-c). Trans-bronchial lung biopsy specimens of right upper and lower lobes revealed thickening of alveolar interstitium with inflammatory cells infiltration without non-caseating epithelioid cell granulomas (Fig. 4d). The PAS-positive lipoproteinaceous material was not observed in the alveolar space. The biopsy specimen of the left finger skin showed increased amounts of collagen fiber in the dermis and atrophy of sweat glands, which were consistent with scleroderma (data not shown). The patient met the 2013 Classification Criteria for Systemic Sclerosis (American College of Rheumatology/ European league against rheumatism collaborative initiative) (van den Hoogen et al. 2013): sclerodermatous change of skin, telangiectasia, Raynaud’s phenomenon, and elevated serum level of ARA; total 12 points/over 9 points. She also met the criteria of sarcoidosis described in the statement on sarcoidosis by ATS, American Thoracic Society, ERS, the European Respiratory Society and WASOG, the World Association of Sarcoidosis and Other Granulomatous Disorders (1999): compatible clinical picture, histologic demonstration of non-caseating granulomas, and exclusion of other diseases capable of producing similar histologic or clinical picture. Therefore, we diagnosed her as having both sarcoidosis and systemic sclerosis. We started the oral prednisolone (PSL) therapy with the dose of 25 mg/day and intravenous cyclophosphamide (IVCY) with 500 mg/4 weeks from February 2014. Her symptoms gradually improved including blurred vision, muscle weakness, and Raynaud’s phenomenon, whereas exertional dyspnea was not improved. The abnormal CT findings were also improved, except for esophageal dilation (Fig. 2d) and interstitial pneumonia in the bilateral lower lobes. We gradually tapered the dose of PSL by 5 mg/day every four weeks and administered six courses of IVCY. The dose of PSL was maintained with 7.5 mg/day, and tacrolimus with the dose of 3 mg/day was started after IVCY. Her symptoms were stable for a while; however, exertional dyspnea worsened and she began to complain of productive cough in March 2015.
She was admitted to our hospital again for close examination. Her temperature was 36.7°C, pulse rate 112 beats/min, respiratory rate 20 breaths/min, and SpO2 93% at room air. MMT was almost normal. The sclerodermatous changes of the skin were observed. Raynaud’s phenomenon and telangiectasia had disappeared. Fine crackles were also heard in bilateral lower fields. Serum levels of AST, ALT, and CPK were normal: 30 IU/L, 14 IU/L, and 53 IU/L, respectively. The serum level of KL-6 was elevated to 11,309 U/mL (Fig. 1). The serum level of ANA was elevated to x160. The other specific auto-antibodies were not elevated. Serum levels of ACE, sIL-2R, and lysozyme were within normal range. The serum level of CEA was elevated to 6.6 ng/mL. Blood tests for infectious disease were all negative, including serum β-D-glucan, Aspergillus-galactomannan antigen, Cryptococcus-antigen, Mycobacterium avium-complex-glycopeptidolipid antibodies, and interferon-gamma release assays. The arterial blood gas analysis detected the decrease of PaO2 to 48 torr at room air. PFT revealed the decrease of %VC and %DLco to 72.8% and 25.0%, respectively. %FEV1 was within normal range (70.5%). A chest CT scan detected a newly-emerged crazy-paving appearance in the bilateral upper and middle lobes (Fig. 2e) and a small nodule with the diameter of 12 mm in the left S9. The appearance of BALF from the left B3b was milky. A microscopic examination of BALF revealed large and foamy macrophages and acellular PAS stain-positive bodies in a diffuse background of granular material (Fig. 4e). A cytological examination of the BALF did not detect any malignant cells, bacteria, or fungi. A cultivation test of the BALF also did not detect any bacteria or fungi. The titer of anti-GM-CSF antibodies measured by ELISA method (Schoch et al. 2002; Uchida et al. 2007) was 10.8 µg/mL (cut off value < 1.0 µg/mL, Fig. 1). Retrospective measurement of serum anti-GM-CSF antibodies, using stocked serum, showed the elevated level of 35.1µg/mL on first admission (Fig. 1). We thus diagnosed her as also having autoimmune PAP according to the following diagnostic criteria: cytological analysis of BALF or pulmonary histopathological findings, chest CT findings, and elevation of serum anti-GM-CSF antibodies levels (≥ 1.0 µg/mL) (Kitamura et al. 2000; Uchida et al. 2014). Six months after the diagnosis of autoimmune PAP, we administered the wedge resection of the left lower lobe for the enlargement of a small nodule with the diameter of 17 mm in the left S9, which was first detected by a chest CT scan on second admission. Pathological findings demonstrated the small nodules as adenocarcinoma. Acellular PAS stain-positive bodies were observed within alveolar spaces, which is consistent with PAP (Fig. 4f).
We tapered the dose of oral PSL to 5 mg/day and stopped the administration of tacrolimus. We repeated segmental BAL (using 500 mL/time of saline, every 3 day-1.5 week) for five courses against the right upper lobe, middle lobe, left superior segment, left lingular segment, and right S8, because the patient refused to receive a whole lung lavage (WLL) under general anesthesia. Her productive cough and exertional dyspnea improved gradually. A chest CT scan showed the improvement of the crazy-paving appearance (Fig. 2f). The serum level of KL-6 decreased to 5,436 U/mL (Fig. 1). The arterial blood gas analysis detected the improvement of PaO2 to 66 torr at room air. PFT showed improvement of %DLco to 45.2%. The %VC and %FEV1 did not change: 75.2% and 69.7%, respectively.
Discussion
Autoimmune PAP rarely accompanies collagen disease or sarcoidosis (Seymour and Presneill 2002; Inoue et al. 2008). However, similar to our case, two patients with collagen disease developed autoimmune PAP during the immunosuppressant therapy (Nagasawa et al. 2013; Imura et al. 2016). On the other hand, two patients developed autoimmune PAP without immunosuppressant therapy, followed by collagen disease or sarcoidosis (Kinehara et al. 2014; Boerner et al. 2016). These results suggest that immunosuppressant therapy could trigger the development of autoimmune PAP in a subset of patients with collagen disease.
Measurement of serum anti-GM-CSF antibodies is used for the diagnosis of autoimmune PAP (Kitamura et al. 2000; Uchida et al. 2014). Serum KL-6 could be the useful marker for not only the disease activity of pulmonary fibrosis (Kohno et al. 1989), but also for the monitoring of autoimmune PAP (Inoue et al. 2006; Lin et al. 2008; Bonella et al. 2013). In our case, at the time of the diagnosis of sarcoidosis and systemic sclerosis, the chest CT scan, BAL, and trans-bronchial lung biopsy could not show the existence of autoimmune PAP; however, serum KL-6 and anti-GM-CSF antibodies levels were already highly elevated. After the immunosuppressant therapy, the serum KL-6 level was further elevated and serum anti-GM-CSF antibodies decreased but remained still high. The chest CT, BAL, and the surgical lung specimen demonstrated the existence of autoimmune PAP. Autoimmune PAP might be in the potential state to develop at the time of diagnosis of sarcoidosis and systemic sclerosis, and immunosuppressant therapy could trigger the onset of autoimmune PAP. Nara et al. (2006) reported a spontaneously remitted case of autoimmune PAP whose serum KL-6 level normalized, whereas serum anti-GM-SCF antibody levels remained high after remission. These results would suggest that the serum anti-GM-SCF antibody is a useful marker for the diagnosis, but not for follow-up of the clinical course; on the other hand, serum KL-6 is an excellent disease activity marker to monitor disease activity of autoimmune PAP. Moreover, serum anti-GM-CSF antibodies should be measured in the case of collagen disease with a very high level of serum KL-6 to exclude the possibility of development of autoimmune PAP during the treatment of steroid or immunosuppressant therapy.
Secondary PAP is caused by macrophage dysfunction due to relative deficiency of GM-CSF that occurs as a consequence of hematological disorders, inhalation injuries, infection, autoimmune diseases, or pharmacologic immunosuppression (Carey and Trapnell 2010). If we did not measure the concentration of serum anti-GM-CSF antibodies, we might diagnose this patient as having secondary PAP, because of the underlying autoimmune disease and usage of immunosuppressant therapy. One of the clinical differences between autoimmune PAP and secondary PAP is a higher frequency of the crazy-paving appearance in chest CT in autoimmune PAP (Ishii et al. 2009). As well as in our case, the finding of the crazy-paving appearance in chest CT might be a clue to measure serum anti-GM-CSF antibodies to distinguish autoimmune PAP from secondary PAP.
The mechanism of how the collagen disease or sarcoidosis co-exists with autoimmune PAP is unknown. The serum autoantibodies against GM-CSF are normally present in healthy people at low levels, and more than 99% of the GM-CSF were bound and neutralized by the GM-CSF autoantibodies (Uchida et al. 2009). In patients with autoimmune PAP, the levels of serum anti-GM-CSF antibodies increase beyond the critical threshold (Carey and Trapnell 2010). Plasma GM-CSF levels were elevated from patients with localized scleroderma (Torok et al. 2015). GM-CSF production by alveolar macrophage in sarcoidosis patients was significantly increased compared with controls (Oltmanns et al. 2003). In our case, the co-existence of systemic sclerosis and sarcoidosis could increase the production of GM-CSF, and then anti-GM-CSF antibodies level might be elevated to neutralize increased GM-CSF. Immunosuppressant therapy decreased the serum anti-GM-CSF antibodies level; however, the level was still high at the onset of autoimmune PAP. Immunosuppressant therapy also might decrease GM-CSF further than the decrease of anti-GM-CSF antibodies. The imbalance between GM-CSF and anti-GM-CSF antibodies might result in the development of autoimmune PAP.
The therapeutic strategy for autoimmune PAP still is not established. WLL is the traditional therapy for PAP (Campo et al. 2016). According to the pathogenesis of autoimmune PAP, we have other therapeutic options: inhalation or subcutaneous administration of GM-CSF (Tazawa et al. 2010; Khan et al. 2012), Rituximab (Borie et al. 2009), and plasmapheresis (Luisetti et al. 2009). Furthermore, the therapeutic strategy against autoimmune PAP associated with collagen disease or sarcoidosis is not totally established. One case of autoimmune PAP associated with systemic lupus erythematosus was successfully treated with a combination of Rituximab, WLL, and the reduction of immunosuppressant therapy (Nagasawa et al. 2013). The other case of autoimmune PAP, followed by dermatomyositis, was successfully remitted only by the reduction of corticosteroid therapy (Imura et al. 2016). Our case could be successfully treated with a combination of immunosuppressant therapy reduction and repeated segmental BAL. Immunosuppressant therapy could be the exacerbation factor in patients with autoimmune PAP (Akasaka et al. 2015). Reducing the dose of immunosuppressant therapy would be the first choice for the treatment of autoimmune PAP associated with collagen disease to restore phagocytic activity of alveolar macrophage and GM-CSF production from GM-CSF-producing cells. The second choice would be the alveolar lavage (WLL or repeated segmental BAL) or immunomodulating therapy (subcutaneous GM-CSF, inhalation of GM-CSF, Rituximab, or plasmapheresis). Further experiences of cases are needed to understand the pathogenesis and establish a therapeutic strategy from patients with autoimmune PAP associated with collagen disease or sarcoidosis.
Acknowledgments
The authors would like to thank Dr. Koh Nakata, Niigata University, for measuring serum anti-GM-CSF antibody levels.
Conflict of Interest
The authors declare no conflict of interest.
References
-
Akasaka,
K.,
Tanaka,
T.,
Kitamura,
N.,
Ohkouchi,
S.,
Tazawa,
R.,
Takada,
T.,
Ichiwata,
T.,
Yamaguchi,
E.,
Hirose,
M.,
Arai,
T.,
Nakano,
K.,
Nei,
T.,
Ishii,
H.,
Handa,
T.,
Inoue,
Y. &
Nakata,
Y.
(2015) Outcome of corticosteroid administration in autoimmune pulmonary alveolar proteinosis: a retrospective cohort study. BMC Pulm. Med., 15, 88.
-
American Thoracic Society (ATS), the European Respiratory Society (ERS) and the World Association of Sarcoidosis and Other Granulomatous Disorders (WASOG)
(1999) Statement on sarcoidosis. Joint Statement of the American Thoracic Society (ATS), the European Respiratory Society (ERS) and the World Association of Sarcoidosis and Other Granulomatous Disorders (WASOG) adopted by the ATS Board of Directors and by the ERS Executive Committee, February 1999. Am. J. Respir. Crit. Care Med., 160, 736-755.
-
Boerner,
E.B.,
Costabel,
U.,
Wessendorf,
T.E.,
Theegarten,
D.,
Hetzel,
M.,
Drent,
M. &
Bonella,
F.
(2016) Pulmonary alveolar proteinosis: another autoimmune disease associated with sarcoidosis? Sarcoidosis Vasc. Diffuse Lung Dis., 33, 90-94.
-
Bonella,
F.,
Ohshimo,
S.,
Miaotian,
C.,
Griese,
M.,
Guzman,
J. &
Costabel,
U.
(2013) Serum KL-6 is a predictor of outcome in pulmonary alveolar proteinosis. Orphanet J. Rare Dis., 8, 53.
-
Borie,
R.,
Debray,
M.P.,
Laine,
C.,
Aubier,
M. &
Crestani,
B.
(2009) Rituximab therapy in autoimmune pulmonary alveolar proteinosis. Eur. Respir. J., 33, 1503-1506.
-
Campo,
I.,
Luisetti,
M.,
Griese,
M.,
Trapnell,
B.C.,
Bonella,
F.,
Grutters,
J.,
Nakata,
K.,
Van Moorsel,
C.H.,
Costabel,
U.,
Cottin,
V.,
Ichiwata,
T.,
Inoue,
Y.,
Braschi,
A.,
Bonizzoni,
G.,
Iotti,
G.A., et al.
(2016) Whole lung lavage therapy for pulmonary alveolar proteinosis: a global survey of current practices and procedures. Orphanet J. Rare Dis., 11, 115.
-
Carey,
B. &
Trapnell,
B.C.
(2010) The molecular basis of pulmonary alveolar proteinosis. Clin. Immunol., 135, 223-235.
-
Imura,
Y.,
Yukawa,
N.,
Handa,
T.,
Nakashima,
R.,
Murakami,
K.,
Yoshifuji,
H.,
Ohmura,
K.,
Ishii,
H.,
Nakata,
K. &
Mimori,
T.
(2016) Two cases of autoimmune and secondary pulmonary alveolar proteinosis during immunosuppressive therapy in dermatomyositis with interstitial lung disease. Mod. Rheumatol., doi.org/10.3109/14397595.2016.1153443 [Epub ahead of print].
-
Inoue,
Y.,
Nakata,
K.,
Arai,
T.,
Tazawa,
R.,
Hamano,
E.,
Nukiwa,
T.,
Kudo,
K.,
Keicho,
N.,
Hizawa,
N.,
Yamaguchi,
E.,
Eda,
R.,
Oishi,
K.,
Maeda,
Y.,
Koreeda,
Y.,
Kodo,
N. &
Sakatani,
M.
(2006) Epidemiological and clinical features of idiopathic pulmonary alveolar proteinosis in Japan. Respirology, 11, Suppl S55-60.
-
Inoue,
Y.,
Trapnell,
B.C.,
Tazawa,
R.,
Arai,
T.,
Takada,
T.,
Hizawa,
N.,
Kasahara,
Y.,
Tatsumi,
K.,
Hojo,
M.,
Ichiwata,
T.,
Tanaka,
N.,
Yamaguchi,
E.,
Eda,
R.,
Oishi,
K.,
Tsuchihashi,
Y., et al.
(2008) Characteristics of a large cohort of patients with autoimmune pulmonary alveolar proteinosis in Japan. Am. J. Respir. Crit. Care Med., 177, 752-762.
-
Ishii,
H.,
Trapnell,
B.C.,
Tazawa,
R.,
Inoue,
Y.,
Akira,
M.,
Kogure,
Y.,
Tomii,
K.,
Takada,
T.,
Hojo,
M.,
Ichiwata,
T.,
Goto,
H. &
Nakata,
K.
(2009) Comparative study of high-resolution CT findings between autoimmune and secondary pulmonary alveolar proteinosis. Chest, 136, 1348-1355.
-
Khan,
A.,
Agarwal,
R. &
Aggarwal,
A.N.
(2012) Effectiveness of granulocyte-macrophage colony-stimulating factor therapy in autoimmune pulmonary alveolar proteinosis: a meta-analysis of observational studies. Chest, 141, 1273-1283.
-
Kinehara,
Y.,
Kida,
H.,
Inoue,
Y.,
Hirose,
M.,
Nakabayashi,
A.,
Takeuchi,
Y.,
Hayama,
Y.,
Fukushima,
K.,
Hirata,
H.,
Inoue,
K.,
Minami,
T.,
Nagatomo,
I.,
Takeda,
Y.,
Funakoshi,
T.,
Kijima,
T. &
Kumanogoh,
A.
(2014) Development of microscopic polyangiitis-related pulmonary fibrosis in a patient with autoimmune pulmonary alveolar proteinosis. BMC Pulm. Med., 14, 172.
-
Kitamura,
T.,
Tanaka,
N.,
Watanabe,
J.,
Uchida,
Kanegasaki,
S.,
Yamada,
Y. &
Nakata,
K.
(1999) Idiopathic pulmonary alveolar proteinosis as an autoimmune disease with neutralizing antibody against granulocyte/macrophage colony-stimulating factor. J. Exp. Med., 190, 875-880.
-
Kitamura,
T.,
Uchida,
K.,
Tanaka,
N.,
Tsuchiya,
T.,
Watanabe,
J.,
Yamada,
Y.,
Hanaoka,
K.,
Seymour,
J.F.,
Schoch,
O.D.,
Doyle,
I.,
Inoue,
Y.,
Sakatani,
M.,
Kudoh,
S.,
Azuma,
A.,
Nukiwa,
T., et al.
(2000) Serological diagnosis of idiopathic pulmonary alveolar proteinosis. Am. J. Respir. Crit. Care Med., 162, 658-662.
-
Kohno,
N.,
Kyoizumi,
S.,
Awaya,
Y.,
Fukuhara,
H.,
Yamakido,
M. &
Akiyama,
M.
(1989) New serum indicator of interstitial pneumonitis activity. Sialylated carbohydrate antigen KL-6. Chest, 96, 68-73.
-
Lin,
F.C.,
Chen,
Y.C. &
Chang,
S.C.
(2008) Clinical importance of bronchoalveolar lavage fluid and blood cytokines, surfactant protein D, and Kerbs von Lungren 6 antigen in idiopathic pulmonary alveolar proteinosis. Mayo Clin. Proc., 83, 1344-1349.
-
Luisetti,
M.,
Rodi,
G.,
Perotti,
C.,
Campo,
I.,
Mariani,
F.,
Pozzi,
E. &
Trapnell,
B.C.
(2009) Plasmapheresis for treatment of pulmonary alveolar proteinosis. Eur. Respir. J., 33, 1220-1222.
-
Nagasawa,
J.,
Kurasawa,
K.,
Maezawa,
R.,
Owada,
T.,
Hanaoka,
R. &
Fukuda,
T.
(2013) Systemic lupus erythematosus complicating autoimmune pulmonary alveolar proteinosis that was worsened by immunosuppressive therapy. Lupus, 22, 1060-1063.
-
Nara,
M.,
Sano,
K.,
Ogawa,
H.,
Tamada,
T.,
Nagaoka,
M.,
Okada,
K.,
Watanabe,
M.,
Moriya,
T.,
Miki,
H.,
Nakata,
K.,
Ichinose,
M. &
Hattori,
T.
(2006) Serum antibody against granulocyte/macrophage colony-stimulating factor and KL-6 in idiopathic pulmonary alveolar proteinosis. Tohoku J. Exp. Med., 208, 349-354.
-
Oltmanns,
U.,
Schmidt,
B.,
Hoernig,
S.,
Witt,
C. &
John,
M.
(2003) Increased spontaneous interleukin-10 release from alveolar macrophages in active pulmonary sarcoidosis. Exp. Lung Res., 29, 315-328.
-
Schoch,
O.D.,
Schanz,
U.,
Koller,
M.,
Nakata,
K.,
Seymour,
J.F.,
Russi,
E.W. &
Boehler,
A.
(2002) BAL findings in a patient with pulmonary alveolar proteinosis successfully treated with GM-CSF. Thorax, 57, 277-280.
-
Seymour,
J.F. &
Presneill,
J.J.
(2002) Pulmonary alveolar proteinosis: progress in the first 44 years. Am. J. Respir. Crit. Care Med., 166, 215-235.
-
Tazawa,
R.,
Trapnell,
B.C.,
Inoue,
Y.,
Arai,
T.,
Takada,
T.,
Nasuhara,
Y.,
Hizawa,
N.,
Kasahara,
Y.,
Tatsumi,
K.,
Hojo,
M.,
Ishii,
H.,
Yokoba,
M.,
Tanaka,
N.,
Yamaguchi,
E.,
Eda,
R., et al.
(2010) Inhaled granulocyte/macrophage-colony stimulating factor as therapy for pulmonary alveolar proteinosis. Am. J. Respir. Crit. Care Med., 181, 1345-1354.
-
Torok,
K.S.,
Kurzinski,
K.,
Kelsey,
C.,
Yabes,
J.,
Magee,
K.,
Vallejo,
A.N.,
Medsger,
T. Jr. &
Feghali-Bostwick,
C.A.
(2015) Peripheral blood cytokine and chemokine profiles in juvenile localized scleroderma: T-helper cell-associated cytokine profiles. Semin. Arthritis Rheum., 45, 284-293.
-
Trapnell,
B.C.,
Whitsett,
J.A. &
Nakata,
K.
(2003) Pulmonary alveolar proteinosis. N. Engl. J. Med., 349, 2527-2539.
-
Uchida,
K.,
Beck,
D.C.,
Yamamoto,
T.,
Berclaz,
P.Y.,
Abe,
S.,
Staudt,
M.K.,
Carey,
B.C.,
Filippi,
M.D.,
Wert,
S.E.,
Denson,
L.A.,
Puchalski,
J.T.,
Hauck,
D.M. &
Trapnell,
B.C.
(2007) GM-CSF autoantibodies and neutrophil dysfunction in pulmonary alveolar proteinosis. N. Engl. J. Med., 356, 567-579.
-
Uchida,
K.,
Nakata,
K.,
Carey,
B.,
Chalk,
C.,
Suzuki,
T.,
Sakagami,
T.,
Koch,
D.E.,
Stevens,
C.,
Inoue,
Y.,
Yamada,
Y. &
Trapnell,
B.C.
(2014) Standardized serum GM-CSF autoantibody testing for the routine clinical diagnosis of autoimmune pulmonary alveolar proteinosis. J. Immunol. Methods, 402, 57-70.
-
Uchida,
K.,
Nakata,
K.,
Suzuki,
T.,
Luisetti,
M.,
Watanabe,
M.,
Koch,
D.E.,
Stevens,
C.A.,
Beck,
D.C.,
Denson,
L.A.,
Carey,
B.C.,
Keicho,
N.,
Krischer,
J.P.,
Yamada,
Y. &
Trapnell,
B.C.
(2009) Granulocyte/macrophage-colony-stimulating factor autoantibodies and myeloid cell immune functions in healthy subjects. Blood, 113, 2547-2556.
-
Uchida,
K.,
Nakata,
K.,
Trapnell,
B.C.,
Terakawa,
T.,
Hamano,
E.,
Mikami,
A.,
Matsushita,
I.,
Seymour,
J.F.,
Oh-Eda,
M.,
Ishige,
I.,
Eishi,
Y.,
Kitamura,
T.,
Yamada,
Y.,
Hanaoka,
K. &
Keicho,
N.
(2004) High-affinity autoantibodies specifically eliminate granulocyte-macrophage colony-stimulating factor activity in the lungs of patients with idiopathic pulmonary alveolar proteinosis. Blood, 103, 1089-1098.
-
van den Hoogen,
F.,
Khanna,
D.,
Fransen,
J.,
Johnson,
S.R.,
Baron,
M.,
Tyndall,
A.,
Matucci-Cerinic,
M.,
Naden,
R.P.,
Medsger,
T.A. Jr.,
Carreira,
P.E.,
Riemekasten,
G.,
Clements,
P.J.,
Denton,
C.P.,
Distler,
O.,
Allanore,
Y., et al.
(2013) 2013 classification criteria for systemic sclerosis: an American college of rheumatology/European league against rheumatism collaborative initiative. Ann. Rheum. Dis., 72, 1747-1755.