Abstract
Campomelic dysplasia (CD) is a skeletal dysplasia characterized by shortened and bowed long bones, airway instability, the potential for disorders of sexual differentiation (DSD), and Pierre Robin Sequence (PRS) with cleft palate, midface hypoplasia and laryngotrachemomalacia. CD is caused by alterations in the Sex-determining region of the Y chromosome (SRY)-related-box 9 (SOX9), which has important roles in tissue and sexual differentiation. The SOX9 gene and the enhancer regions of SOX9 are located at chromosome 17q24.3. We report a 6-year-old phenotypically female referred to our department because of precocious puberty. The patient was born with Tetralogy of Fallot (TOF) and PRS. Skeletal X-ray examination showed only 11 pairs of ribs and bilateral bowed radiuses. Endocrine evaluations showed that increased levels of serum testosterone, and chromosomal analysis revealed a 46, XY, t(2;17)(p15;q24.2) karyotype. The patient was diagnosed with peripheral precocious puberty caused by over-secretion of testosterone by gonadoblastoma originating from dysgenetic gonads with Y-chromosome-related DSD. Multiple somatic abnormalities and DSD indicated that the patient might have CD. Laparoscopy revealed bilateral dysgenetic gonads, and these were removed in the successive operation to prevent malignant transformation and virilization, caused by dysgenetic gonads with Y chromosomal materials. It is highly suggestive that the chromosomal translocation of 17q 24.2 may cause DSD and multiple somatic abnormalities, including CD, although the identified 17q breakpoint was located outside of known SOX9 enhancer regions. Thus, a hitherto unknown enhancer may be present at 17q24.2. This is the first reported case of CD with a translocation breakpoint at 17q24.2.
Introduction
Sex-determining region of the Y chromosome (SRY)-related-box 9 (SOX9) plays a pivotal role in sexual differentiation (Walters-Sen et al. 2014). The SOX9 gene and the enhancer regions of SOX9 are located at chromosome 17q24.3 (Walters-Sen et al. 2014). Lack of SOX9 or its missense mutation often results in disorders of sex development (DSD) associated with dysgenetic gonads. Abdominal dysgenetic gonads with Y chromosomal materials can lead to a higher occurrence of gonadoblastoma and malignant tumors (Tam et al. 2016). Moreover, excessive sex-hormone production by gonadoblastoma may also result in peripheral precocity. Therefore, dysgenetic gonads with Y chromosomal materials must be removed as soon as possible after diagnosis (Tam et al. 2016).
SOX9 has two important roles in tissue differentiation. Firstly, it works as an important downstream effector for the SRY gene in sexual differentiation. Secondly, it promotes differentiation of chondrocyte and is associated with development of several organs. This is why SOX9 alteration can lead to multiple malformations including DSD (Walters-Sen et al. 2014).
Campomelic dysplasia (CD) is a rare genetic disorder characterized by bowing of the long bones and many other skeletal and extraskeletal features. An atypical form of the disease with absence of bowed limbs is called acampomelic CD (ACD) and is found in about 10% of patients (Walters-Sen et al. 2014).
Here we report a rare case of 6-year-old phenotypically female with CD, tetralogy of Fallot (TOF), skeletal anomalies and Pierre Robin Sequence (PRS) characterized by cleft palate, midface hypoplasia and laryngotracheomalacia. A chromosomal analysis revealed a 46, XY genotype, as well as a chromosome breakpoint at chromosome 17q24.2 in which the enhancer of SOX9 has never been reported to be present. Presumably, translocation of chromosome 17q 24.2 may be responsible for DSD, PRS, TOF, and CD. In this sense, this is the first report of such a case in the world.
Case Report
A 6-year-and-10-month-old phenotypically normal female was referred to the pediatric endocrinology department complaining of premature thelarche and precocious pubarche that had been observed for the past 9 months. She was born with a palatal cleft due to PRS, and TOF. She had undergone palatoplasty and cardiac surgery in the past.
The patient was presented as a phenotypically normal female. Physical examination showed that she had Tanner grade 3 pubic hair, breast development and female external genitalia with a mild clitoral hypertrophy (Fig. 1). Skeletal X-ray examination showed only 11 pairs of ribs and bilateral bowed radiuses. Any abnormal findings including brain tumor were not observed on cranial magnetic resonance imaging (MRI). An abdominal MRI confirmed a normally developed uterus and vagina; however, the gonads were not identified. Endocrine evaluations showed elevated serum testosterone, while luteinizing hormone (LH), follicle stimulating hormone (FSH), aldosterone, and cortisol levels were within normal ranges (Table 1). The LH-releasing hormone (LH-RH) test showed LH and FSH levels were within reference value. The human chorionic gonadotropin (hCG) test showed further increases of testosterone, and human menopausal gonadotropin (hMG) test showed no effect on estradiol (E2) levels. There was a slight elevation of E2 levels, which was thought to be caused by conversion from excessive testosterone by aromatase. A chromosomal analysis revealed a 46, XY, t(2;17)(p15;q24.2) karyotype (Fig. 2). Based on these results, the patient was diagnosed with peripheral precocity caused by the elevation of testosterone originating from some kind of endocrine disorder. Moreover, her skeletal abnormalities and DSD indicated that she might have CD.
There had been some serious difficulties to make an accurate diagnosis in the endocrine department. Nine months had passed from the first visit to our institution, when she was referred to our department to evaluate her gonads and internal genital organs by laparoscopy. The patient had grown 8.1 cm in height during the 9 months. The voice had become deeper, and the patient had facial and underarm hair. These findings were compatible with virilism, which indicated an excessive production of testosterone. Vaginoscopy showed a matured vagina and internal ostium of the uterus. Laparoscopy also showed a normal uterus, but an oviduct–like structure and abnormally massive white gonads were found on both sides (Fig. 3). Laparoscopic removal of bilateral gonads was performed, as they were diagnoses as dysgenetic gonads. A pathological examination showed that the gonads were testes and most parts had transformed into gonadoblastoma.
After the surgery, testosterone levels were decreased to 5.3 ng/dl, and the symptoms of precocious puberty improved dramatically. The patient’s parents had a strong wish to raise her as a girl.
Discussion
A translocation at chromosome 17q24.2 in the present patient with DSD has never been reported in the literature (Walters-Sen et al. 2014). As SOX9 is the important downstream effector for the SRY gene in sexual differentiation, alteration of SOX9 results in DSD with dysgenetic gonads, which leads to the development of gonadoblastoma or malignant tumor. Gonadoblastoma is a type of hamartoma generated from dysgenetic gonads with Y-chromosome-related DSDs (Kearsey and Hutson 2017). In addition, gonadoblastoma sometimes secretes sex hormones and causes several clinical manifestations such as precocious puberty (Iliev et al. 2002). Hence, to prevent malignant transformation and virilization, dysgenetic gonads with Y chromosomal materials should be removed as soon as possible after the diagnosis is confirmed (Tam et al. 2016).
SOX9 also plays a role in differentiation of cartilage tissue. Alteration of SOX9 causes not only cartilage disorders such as PRS but also skeletal dysplasia such as CD (Zody et al. 2006). There is a correlation between the distance from the chromosome translocation site to the SOX9 locus and the severity of the disease phenotype. ACD is a mild phenotype of CD and is associated with chromosome translocation in areas where the enhancer region of SOX9 is present. The enhancer region exists in up to 2.0 Mb upstream of SOX9, and the chromosome translocation that results in CD or ACD exists at 50-375 kb, 379-459 kb, 585-932 kb, and 1.02-1.20 Mb upstream of SOX9. These are all a part of the locus 17q24.3 (Gordon et al. 2014; Karaer et al. 2014; Kim et al. 2015; Katoh-Fukui et al. 2015; Castori et al. 2016). In the present case, a FISH analysis found that the breakpoint existed at chromosome 17q24.2, which has never been reported with respect to CD (Fig. 4). This is the first case report of CD with a chromosome breakpoint at 17q24.2, suggesting a possibility that the enhancer region may be present in this region upstream from those that have been described in previous reports. On the other hand, we supposed that there was no association between the breakpoint at 2p15 and CD or DSD in this case. There have been no reports about CD related with chromosome 2, although the genes in chromosome 2 related to DSD were reported to be located at 2q31 (Slavotinek et al. 1999). We could not perform further molecular analysis to confirm our speculation, since her parents did not want additional examination. Thus, we cannot exclude the possibility that this patient has a complex structure rather than a simple translocation. Further analysis such as array-based comparative genomic hybridization could provide additional information of this genomic rearrangement.
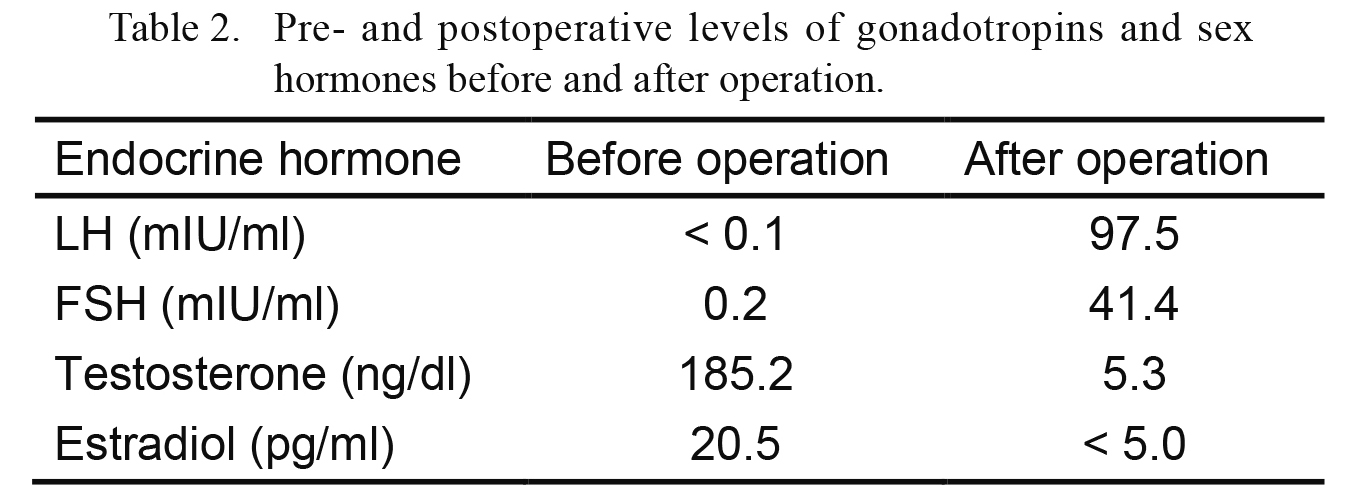
The present patient has multiple malformations, such as PRS and TOF, that could be caused by alteration of SOX9. However, she had never been diagnosed with CD or DSD until signs of precocious puberty began to appear. The presumption is that the decreased expression of SOX9 caused the decreased secretion of Müllerian inhibiting substance (MIS) and testosterone. MIS generated from sertoli cells inhibits the formation of uterus and the oviduct, and testosterone generated from Leydig cells is related to virilization. In this case, the uterus and the oviduct were formed due to the decreased secretion of MIS, and virilization of the external genitalia was inhibited by the insufficient secretion of testosterone. Gonadoblastoma might have developed from the dysgenetic gonads and began to secrete testosterone when the patient was around 6 years old. This hypothesis was proved by the fact that the value of testosterone was decreased while LH and FSH levels were increased after gonadal resection (Table 2).
Gender is a fundamental characteristic of a person. Thus, for patients with DSD, the determination of gender is very important and should not be delayed. This patient was raised as a girl despite having a 46, XY genotype, because she had never been diagnosed with DSD until precocious puberty began to develop. However, she has several SOX9-related malformations. DSD is a rare disease and is often overlooked by clinicians. Therefore, medical staff, including urologists, should always keep DSD in mind when examining a patient with SOX9-related disorders.
This is the first reported case of CD with a translocation breakpoint at 17q24.2. We have proposed the possibility that the hitherto unknown enhancer of SOX9 exists at 17q24.2. Further research in this area is strongly recommended.
Acknowledgments
This study has been approved by ethical review committee in Miyagi Children’s Hospital. Both the patient and parents have agreed to paper submission.
Conflict of Interest
The authors declare no conflict of interest.
References
-
Castori,
M.,
Bottillo,
I.,
Morlino,
S.,
Barone,
C.,
Cascone,
P.,
Pediatric Craniofacial Malformation (PECRAM) Study Group,
Grammatico,
P. &
Laino,
L.
(2016) Variability in a three-generation family with Pierre Robin sequence, acampomelic campomelic dysplasia, and intellectual disability due to a novel ~ 1 Mb deletion upstream of SOX9, and including KCNJ2 and KCNJ16. Birth Defects Res. A Clin. Mol. Teratol., 106, 61-68.
-
Gordon,
C.T.,
Attanasio,
C.,
Bhatia,
S.,
Benko,
S.,
Ansari,
M.,
Tan,
T.Y.,
Munnich,
A.,
Pennacchio,
L.A.,
Abadie,
V.,
Temple,
I.K.,
Goldenberg,
A.,
van Heyningen,
V.,
Amiel,
J.,
FitzPatrick,
D.,
Kleinjan,
D.A., et al.
(2014) Identification of novel craniofacial regulatory domains located far upstream of SOX9 and disrupted in Pierre Robin sequence. Hum. Mutat., 35, 1011-1020.
-
Iliev,
D.I.,
Ranke,
M.B. &
Wollmann,
H.A.
(2002) Mixed gonadal dysgenesis and precocious puberty. Horm. Res., 58, 30-33.
-
Karaer,
K.,
Yuksel,
Z.,
Yalinbas,
E. &
Scherer,
G.
(2014) A case of campomelic dysplasia in whom a new mutation was found in the SOX9 gene. Turk. Pediatri Ars., 49, 154-156.
-
Katoh-Fukui,
Y.,
Igarashi,
M.,
Nagasaki,
K.,
Horikawa,
R.,
Nagai,
T.,
Tsuchiya,
T.,
Suzuki,
E.,
Miyado,
M.,
Hata,
K.,
Nakabayashi,
K.,
Hayashi,
K.,
Matsubara,
Y.,
Baba,
T.,
Morohashi,
K.,
Igarashi,
A., et al.
(2015) Testicular dysgenesis/regression without campomelic dysplasia in patients carrying missense mutations and upstream deletion of SOX9. Mol. Genet. Genomic Med., 3, 550-557.
-
Kearsey,
I. &
Hutson,
J.M.
(2017) Disorders of sex development (DSD): not only babies with ambigious genitalia. A practical guide for surgeons. Pediatr. Surg. Int., 33, 355-361.
-
Kim,
G.J.,
Sock,
E.,
Buchberger,
A.,
Just,
W.,
Denzer,
F.,
Hoepffner,
W.,
German,
J.,
Cole,
T.,
Mann,
J.,
Seguin,
J.H.,
Zipf,
W.,
Costigan,
C.,
Schmiady,
H.,
Rostasy,
M.,
Kramer,
M., et al.
(2015) Copy number variation of two separate regulatory regions upstream of SOX9 causes isolated 46,XY or 46,XX disorder of sex development. J. Med. Genet., 52, 240-247.
-
Slavotinek,
A.,
Schwarz,
C.,
Getty,
J.F.,
Stecko,
O.,
Goodman,
F. &
Kingston,
H.
(1999) Two cases with interstitial deletions of chromosome 2 and sex reversal in one. Am. J. Med. Genet., 86, 75-81.
-
Tam,
Y.H.,
Wong,
Y.S.,
Pang,
K.K.,
To,
K.F.,
Yiu,
A.K.,
Wong,
H.Y.,
Tsui,
S.Y.,
Mou,
J.W.,
Chan,
K.W. &
Lee,
K.H.
(2016) Tumor risk of children with 45,X/46,XY gonadal dysgenesis in relation to their clinical presentations: further insights into the gonadal management. J. Pediatr. Surg., 51, 1462-1466.
-
Walters-Sen,
L.C.,
Thrush,
D.L.,
Hickey,
S.E.,
Hashimoto,
S.,
Reshmi,
S.,
Gastier-Foster,
J.M.,
Pyatt,
R.E. &
Astbury,
C.
(2014) Atypical breakpoint in a t(6;17) translocation case of acampomelic campomelic dysplasia. Eur. J. Med. Genet., 57, 315-318.
-
Zody,
M.C.,
Garber,
M.,
Adams,
D.J.,
Sharpe,
T.,
Harrow,
J.,
Lupski,
J.R.,
Nicholson,
C.,
Searle,
S.M.,
Wilming,
L.,
Young,
S.K.,
Abouelleil,
A.,
Allen,
N.R.,
Bi,
W.,
Bloom,
T.,
Borowsky,
M.L., et al.
(2006) DNA sequence of human chromosome 17 and analysis of rearrangement in the human lineage. Nature, 440, 1045-1049.