Abstract
Epidemiologic studies indicate that exposure to psychosocial stress in early childhood is a risk factor of adult-onset asthma, but the mechanisms of this relationship are poorly understood. Therefore, we examined whether early-life stress increases susceptibility to adult-onset asthma by inhibiting the development of respiratory tolerance. Neonatal BALB/c female mice were aerosolized with ovalbumin (OVA) to induce immune tolerance prior to immune sensitization with an intraperitoneal injection of OVA and the adjuvant aluminum hydroxide. Maternal separation (MS) was applied as an early-life stressor during the induction phase of immune tolerance. The mice were challenged with OVA aerosol in adulthood, and allergic airway responses were evaluated, including airway hyper-responsiveness to inhaled methacholine, inflammatory cell infiltration, bronchoalveolar lavage fluid levels of interleukin (IL)-4, IL-5, and IL-13, and serum OVA-specific IgE. We then evaluated the effects of MS on the development of regulatory T (Treg) cells in bronchial lymph nodes (BLN) and on splenocyte proliferation and cytokine expression. In mice that underwent MS and OVA tolerization, the allergic airway responses and OVA-induced proliferation and IL-4 expression of splenocytes were significantly enhanced. Furthermore, exposure to MS was associated with a lower number of Treg cells in the BLN. These findings suggest that exposure to early-life stress prevents the acquisition of respiratory tolerance to inhaled antigen due to insufficient Treg cell development, resulting in Th2-biased sensitization and asthma onset. We provide the evidence for inhibitory effects of early-life stress on immune tolerance. The present findings may help to clarify the pathogenesis of adult-onset asthma.
Introduction
Bronchial asthma is estimated to affect more than 315 million people worldwide, making it the most common chronic inflammatory disease (To et al. 2012). Asthma is characterized by airway inflammation leading to airway hyper-responsiveness and tissue remodeling and has been shown to be related to various environmental factors, including allergens, infection, and psychosocial stress. The asthmatic inflammatory response is caused by eosinophil infiltration, increased mucous production, and elevated serum IgE levels, which are in turn orchestrated by type-2 T helper (Th2) cells and Th2 cytokines such as interleukin (IL)-4, IL-5, and IL-13. Th2 immune responses are negatively regulated by type-1 T helper (Th1) immune responses and immune tolerance in which regulatory T (Treg) cells play a central role (Fahy 2015; Holgate et al. 2015; Lambrecht and Hammad 2015; Kim et al. 2016; Rothenberg et al. 2017).
Treg cells play a critical role in maintaining immune network balance by suppressing or limiting effector immune responses against internal and external insults, including preventing organ-specific autoimmunity, preventing allograft rejection, and maintaining self-tolerance (Palomares et al. 2010; Sakaguchi et al. 2010; Peterson 2012; Martin-Orozco et al. 2017). Generation of Treg cells appears to be dependent on microenvironmental factors, including IL-10 and transforming growth factor β (Palomares et al. 2014; Noval Rivas and Chatila 2016). Compared to healthy subjects, severe asthma patients show reduced numbers of Treg cells in the peripheral blood and sputum as well as impaired suppressive activities (Mamessier et al. 2008). Moreover, the percentage of Treg cells in the bronchoalveolar lavage fluid (BALF) is lower in children (Hartl et al. 2007) and adults (Barczyk et al. 2014) with asthma than in healthy subjects. The failure of Treg cells to suppress Th2-mediated immune responses to innocuous inhaled antigens may lead to an imbalance between the responses of Treg cells and effector Th2 cells to allergens, resulting in the development and maintenance of asthmatic airway inflammation characterized by an uncontrolled Th2-biased immune response (Lloyd and Hawrylowicz 2009; Fujita et al. 2012; Noval Rivas and Chatila 2016).
Recent studies have investigated the mechanisms underlying the impairment of immune tolerance associated with increased susceptibility to allergic asthma. It has been shown in a mouse model that respiratory infections with viruses such as rhinovirus and respiratory syncytial virus (RSV) in early life are risk factors for developing asthma in adult life. Simultaneous rhinovirus infection and tolerance induction in mice inhibited the acquisition of immune tolerance, attenuating the development of Treg cells characterize with the expression of a specific transcription factor, Foxp3 (Mehta et al. 2016). RSV infection impaired immune tolerance by inducing phenotypic changes in Foxp3+ Treg cells in an IL-4 dependent pathway, including expression of Th2-specefic transcription factor, GATA-3, Th2 cytokine production, and loss of suppressive functions (Krishnamoorthy et al. 2012). IL-33 exposure evoked Th2-like properties in Foxp3+ Treg cells, resulting in airway inflammation in tolerized mice (Chen et al. 2017). Further, suppression of prostaglandin I2 receptor signaling by administration of indomethacin augmented allergen-induced airway inflammation in tolerized mice without affecting Treg cells levels (Zhou et al. 2014). These results suggest that impaired respiratory tolerance leading to increased susceptibility to allergic asthma may result from multiple mechanisms and causes. However, the mechanisms by which tolerogenic immune responses are impaired in asthmatic patients remain to be explored.
Early-life stress exposure has been associated with an increased risk of adult-onset asthma (Ilmarinen et al. 2015; Rosa et al. 2018). For example, experiences of abuse during childhood and adolescence have been shown to be positively associated with adult-onset asthma in African American women (Coogan et al. 2013). Furthermore, childhood maltreatment was associated with elevated odds of lifetime diagnosis of asthma (Scott et al. 2012). However, the pathogenic mechanism underlying the relationship between adult-onset asthma development and early-life stress is unknown.
Various kinds of stressors have been reported to decrease Treg cell production. The number of Treg cells in the peripheral blood of patients with post-traumatic stress disorder has been shown to be significantly lower than that in control subjects (Loerbroks et al. 2009; Jergovic et al. 2014). Further, an acute laboratory stressor was able to decrease the number of CD4+Foxp3+ Treg cells in the peripheral blood of human subjects (Freier et al. 2010). Our group previously demonstrated that the use of restraint as a psychological stressor in adulthood increased susceptibility to asthma by inhibiting the development of Treg cells in a murine model of allergic asthma (Kawano et al. 2018).
Therefore, we have hypothesized that exposure to psychological stress in early life prevents the development of respiratory tolerance by inhibiting Treg cell generation, resulting in a Th2-biased immune response followed by increased susceptibility to allergic asthma. To investigate this hypothesis, we examined the effects of early-life stress on the induction of respiratory tolerance in a murine model of allergic asthma.
Materials and Methods
Mice
Specific-pathogen-free 4-postnatal day (PND)-old female BALB/c mice were purchased with their dams from CLEA Japan (Tokyo, Japan). Mice were housed under a 12-h light/dark cycle at a constant temperature. Sterilized food and water were available ad libitum. All experiments described below were approved by the Committee of Animal Experiments at Tohoku Medical and Pharmaceutical University (approval numbers: 14002-cn, 15001-cn, 16002-cn and 17004-cn). Further, all experiments were performed in compliance with relevant institutional guidelines. We took the utmost care to alleviate any pain and suffering in the mice.
Maternal separation
To model early-life stress, mice were subjected to maternal separation (MS) for 3 h per day for 6 consecutive days on PNDs 17-22 (MS mice). As a control for the stress exposure, MS was not performed for a subset of the mice (non-MS mice). Dams were removed from their home cages and placed in identical new cages until the end of the separation period. Each mouse was isolated in a separate container. At the end of the separation period, mice were returned to their home cages, followed by reunion with their dams. In humans, stress caused by early-life adverse events, such as parental neglect, is often associated with disorders of the hypothalamic-pituitary-adrenal (HPA) axis leading to unexpected stress responses including increased anxiety and depression (Tarullo and Gunnar 2006; Maniam et al. 2014). Also, in animal models, early-life stress, such as maternal separation, has been well known to result in reprogramming of the HPA axis associated with increased anxiety-like behavior and exaggerated stress responsiveness in adulthood (Murgatroyd and Spengler 2011; Nishi et al. 2014), and maternal separation is most commonly used as early-life stress in murine models (Tractenberg et al. 2016). Therefore, maternal separation in murine models would be feasible for the early-life stress in childhood of human beings.
Protocols for stress exposure, tolerization, sensitization, and antigen challenge
The mice were sensitized and made to inhale antigen, as described previously (Kawano et al. 2018), after stress exposure and tolerization (Fig. 1). Respiratory tolerance was induced by chicken ovalbumin (OVA) (grade V; Sigma-Aldrich, St. Louis, MO) inhalation on PNDs 18 and 21 with or without MS as described above. For 30 min before stress exposure, aerosolized OVA (5 mg/ml of saline) was flowed through the container (tolerized mice). As a control for tolerance, saline alone was flowed through the containers for a subset of the mice (non-tolerized mice). Then, all mice were ablactated on PND 23, and all 4 groups of mice (non-MS/non-tolerized group, non-MS/tolerized group, MS/non-tolerized group and MS/tolerized group) were subjected to sensitization. Briefly, OVA (8 µg/mouse) adsorbed onto aluminum hydroxide (Wako Pure Chemical Industries, Osaka, Japan) (4 mg/mouse) was intraperitoneally injected into each mouse on PNDs 24 and 29. On PND 76, all mice were challenged with aerosolized OVA or saline. On PND 77 and PND 81, bronchoalveolar lavage fluids (BALFs) were obtained (Kawano et al. 2018) and processed to measure cytokine content, total cell counts, and cell differentials. After centrifugation, BALF supernatants were stored at –80°C before performing enzyme-linked immunosorbent assays (ELISAs) to measure cytokine levels. Lung tissues were processed for the evaluation of mucus secretion and peribronchial inflammation.

Before and immediately after MS-stress exposure on PND 17 and immediately after MS-stress exposure on PNDs 19 and 22, blood was collected from stressed and non-stressed mice as previously described (Kawano et al. 2018). Plasma samples separated from blood were stored at −80°C for corticosterone concentration measurements using an enzyme immunoassay kit (Enzo Life Sciences, Farmingdale, NY) according to the manufacturer’s protocol. The sensitivity of detection was 32 pg/ml.
Histological analysis of the lungs
Sections of lung tissues collected on PND 81 were stained with periodic acid-Schiff (PAS) to evaluate mucus-containing goblet cells in the bronchial epithelium or with hematoxylin and eosin to study eosinophil infiltration in the peribronchial areas. The number of PAS-positive cells and eosinophils were determined as described previously (Kawano et al. 2018).
OVA-specific IgE measurement
The concentrations of OVA-specific IgE in sera collected on PND 81 were measured with ELISA, as described previously (Kawano et al. 2018). The sensitivity of detection was 1.9 × 10−2 EU/ml for IgE.
Culture of splenocytes
Splenocytes were prepared from mice on PND 76 before the challenge. The cells were cultured at 1 × 106 cells in 200 µl/well in 96-well culture plates in the presence or absence of OVA (200 μg/ml) for 72 h. The samples were then centrifuged, and the supernatants were stored at −80°C for subsequent cytokine measurements. Cell proliferation was measured as previously described (Kawano et al. 2018).
Preparation of bronchial lymph node cells
Bronchial lymph nodes (BLNs) obtained from ten non-toleraized mice and from 3 toleraized mice with or without the stress exposure after the last MS on PND 22 were pooled and processed for the preparation of BLN cells as previously reported (Kawano et al. 2018). BLN cells were then analyzed with a flow cytometer as described below.
Flow-cytometric analysis
Cells were pre-incubated with anti-CD16/CD32 (FC gamma III/II receptor; BD Biosciences, Franklin Lanes, NJ) in order to reduce nonspecific binding of subsequent antibodies. Dead cells were excluded using a LIVE/DEAD Fixable Blue Dead Cell Stain Kit (Thermo Fisher Scientific, Waltham, MA). The cells were stained for surface antigens with anti-CD3ε-FITC (clone 145-2C11; Miltenyi Biotec, Bergisch Gladbach, Germany), anti-CD4-AF700 (clone RM4-5; BD Biosciences), or isotype-control antibodies. For intracellular staining, cells were stimulated with phorbol 12-myristate 13-acetate (PMA) (5 ng/ml), ionomycin (500 ng/ml), and monensin (2 µM) for 4 h before surface-antigen staining. After fixation and permeabilization, the cells were incubated with anti-Foxp3-PE-Cy7 (clone FJK-16s; Thermo Fisher Scientific) or an isotype-control antibody. We considered CD3+CD4+Foxp3+ cells to be Treg cells. Cells were counted on a FACSAria II flow cytometer (BD Biosciences), and the analyses were performed using FACSDiva software (BD Biosciences).
Measurements of cytokine contents
The concentrations of IL-4, IL-5, IL-13 and IFN-γ in the BALF supernatants and the splenocyte culture supernatants were measured using ELISA kits (Thermo Fisher Scientific) according to the manufacturer’s protocol. The sensitivity of detection was 2 pg/ml for IL-4, 7 pg/ml for IL-5, 1.5 pg/ml for IL-13, and 8 pg/ml for IFN-γ.
Statistical analysis
Data are expressed as mean ± standard deviation (SD) from multiple independent experiments (as indicated by n values). Significant differences between pairs of groups were determined by the nonparametric Mann-Whitney U-test. For comparisons between multiple groups, analysis of variance and Bonferroni’s post-hoc tests were performed. These analyses were performed using Prism6 (GraphPad Software, San Diego, CA). P < 0.05 was considered statistically significant.
Results
Effects of maternal separation on plasma corticosterone concentrations
Blood was collected from stressed and non-stressed mice before and immediately after the 1st MS-stress exposure on PND 17 and immediately after the 3rd and 6th MS-stress exposures on PNDs 19 and 22. The mean corticosterone concentrations (ng/ml) in MS mice were significantly higher than those in non-MS mice at all 3 of the measured time points (1st: 185.2 ± 191.1 [n = 6] vs. 55.9 ± 38.1 [n = 6], P < 0.05; 3rd: 267.8 ± 234.9 [n = 6] vs. 106.4 ± 83.9 [n = 7], P < 0.05; 6th: 122.3 ± 168.7 [n = 7] vs. 24.1 ± 19.8 [n = 6], P < 0.05). There were no significant differences in corticosterone concentrations between MS and non-MS mice before the stress exposures (Fig. 2).
Effects of MS on airway responsiveness
To investigate the effects of early life stress on asthmatic airway responses, we first observed airway hyper-responsiveness to inhaled methacholine. As expected, non-MS/tolerized mice (n = 7) showed significantly lower airway reactivity (RL) than non-MS/non-tolerized mice (n = 6) at methacholine concentrations of 0.625 mg/mL (1.37 ± 0.21 vs. 2.01 ± 0.57, P < 0.05), 1.25 mg/mL (1.57 ± 0.38 vs. 3.37 ± 1.24, P < 0.01), 2.5 mg/mL (1.92 ± 0.46 vs. 6.79 ± 2.25, P < 0.01), and 5 mg/mL (3.17 ± 1.2 vs. 6.85 ± 1.96, P < 0.01) (Fig. 3A). Similarly, airway sensitivity in non-MS/tolerized mice was significantly lower, shown by higher PC200, than in non-MS/non-tolerized mice (3.60 ± 0.87 vs. 1.18 ± 0.25, P < 0.01) (Fig. 3B). Interestingly, MS completely abolished the inhibitory effects of tolerization on airway hyper-responsiveness. Both airway reactivity and airway sensitivity were significantly higher in MS/tolerized mice (n = 5) than in non-MS/tolerized mice (RL, 1.25 mg/mL of methacholine: 3.15 ± 1.85 vs. 1.57 ± 0.38, P < 0.05; 2.5 mg/mL: 7.80 ± 4.24 vs. 1.92 ± 0.46, P < 0.01; 5 mg/mL: 6.13 ± 3.28 vs. 3.17 ± 1.20, P < 0.05; Fig. 3A; PC200; 1.44 ± 0.61 vs. 3.60 ± 0.87, P < 0.01; Fig. 3). Further, both of these measures in the MS/tolerized mice (n = 5) were statistically similar to those in the MS/non-tolerized mice (n = 3; Fig. 3). There were no significant differences in RL and PC200 between non-MS/non-tolerized mice and MS/non-tolerized mice, suggesting that MS affects airway hyper-responsiveness by modifying the effects of tolerization.

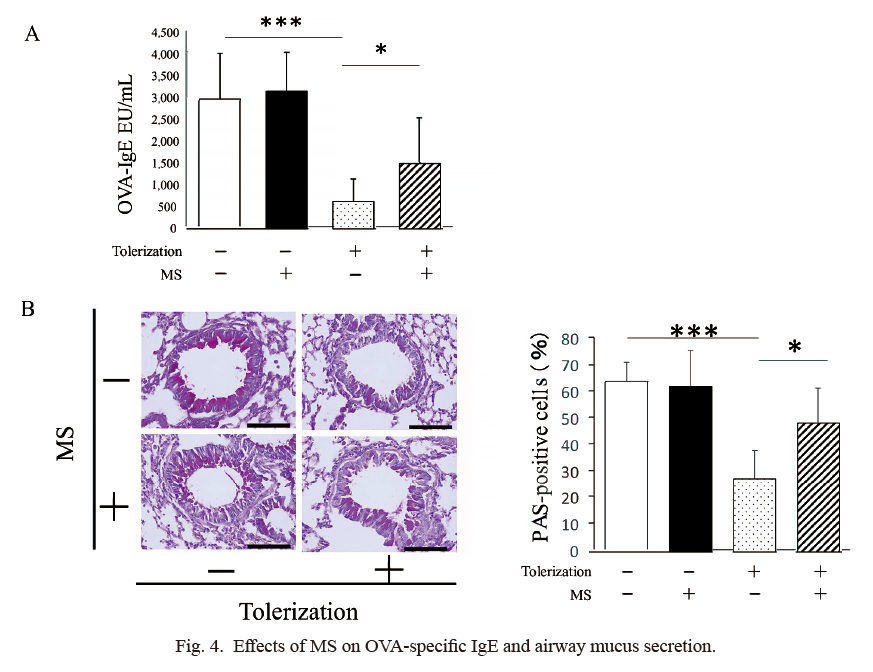
Five days after the OVA challenge (PND 81), the amount of OVA-specific IgE was significantly lower in non-MS/tolerized mice than in non-MS/non-tolerized mice (627.2 ± 519.6 EU/ml [n = 8] vs. 2,987.8 ± 1,049.7 EU/ml [n = 8], P < 0.001). Further, the IgE levels in the MS/tolerized mice were significantly higher than the levels in the non-MS/tolerized mice (1,501.3 ± 1,004.0 EU/ml [n = 6] vs. 627.2 ± 519.6 EU/ml [n = 8], P < 0.05). Additionally, the IgE levels in the MS/tolerized mice were statistically similar to those in the MS/non-tolerized mice. There was no significant difference in the IgE levels between the non-MS/non-tolerized mice and the MS/non-tolerized mice (Fig. 4A). The numbers of PAS-positive cells in the non-MS/tolerized mice were significantly lower than those in the non-MS/non-tolerized mice, and the numbers in the MS/tolerized mice were significantly lower than those in the non-MS/tolerized mice (48.6 ± 12.2% [n = 6] vs. 27.6 ± 10.6% [n = 8], P < 0.05). The number of PAS-positive cells in the MS/tolerized mice was statistically similar to that in the MS/non-tolerized mice. No significant difference in the number of PAS cells was observed between the non-MS/non-tolerized mice and the MS/non-tolerized mice (Fig. 4B). These findings, shown in Fig. 4, suggest that MS affects OVA-specific IgE levels and mucus secretion by modifying the effects of tolerization.
As expected, the non-MS/tolerized mice showed significantly lower numbers of total cells, eosinophils, and lymphocytes in BALF (Fig. 5) compared with the non-MS/non-tolerized mice. OVA-specific IgE and mucus secretion were each measured to assess airway hyper-responsiveness, and the numbers of those cell types in BALF were significantly increased in the MS/tolerized mice compared with the non-MS/tolerized mice. Interestingly, MS significantly increased the number of neutrophils in tolerized, but not non-tolerized, mice. Similar effects of MS were also observed in the Th2 cytokine contents of the BALF (Fig. 6). The BALF concentrations of IFN-γ were not significantly different between the 4 groups. No effects of MS on airway inflammation or Th2 cytokine contents were observed when comparing MS/non-tolerized mice with non-MS/non-tolerized mice. These data indicate that MS modifies the effects of respiratory tolerization.
Effects of MS on sensitization in tolerized mice
Because MS led to the development of antigen-induced asthmatic responses in tolerized mice, we also investigated the effects of MS on immune tolerance. As expected, pretreating mice with aerosolized-OVA inhibited splenocyte proliferation upon OVA stimulation in vitro. However, in vitro splenocyte proliferation was significantly higher in the mice that were exposed to MS during the induction phase of tolerance compared to the non-MS tolerized mice. Indeed, the MS/tolerized mice and the non-tolerized mice showed similar levels of in vitro splenocyte proliferation. MS had no effects on splenocyte proliferation under non-tolerized condition (Fig. 7A). Moreover, we compared OVA-induced cytokine expression (IL-4 and IFN-γ) by splenocytes in all 4 groups (Fig. 7B and C). Tolerization significantly decreased IL-4 expression in non-MS mice, while MS exposure significantly increased cytokine expression in tolerized mice. The MS/tolerized mice and the non-tolerized mice showed similar IL-4 levels (Fig. 7B). Tolerization had no effects on IFN-γ regardless of whether the mice were exposed to MS or not (Fig. 7C). MS had no effect on cytokine expression in the non-tolerized mice.

Because MS inhibited the induction of respiratory tolerance, we analyzed CD3+CD4+Foxp3+ cells as Treg cells in BLNs. The percentage of Treg cells was significantly increased in tolerized mice compared with non-toleraized mice under the non-MS condition. Then, the MS exposure significantly decreased percentage of Treg cells in MS/tolerized mice compared with non-MS/tolerized mice. The percentage of Treg cells in MS/non-tolerized mice was statistically similar to that in non-MS/non-tolerized mice, suggesting that MS had no noticeable effects on the induction of Treg cells under non-tolerized condition. Additionally, the percentage of Treg cells was not statistically different between non-tolerized and tolerized mice under the MS condition (Fig. 8). Together with those findings, we suggest that MS could affect the induction of Treg cells by modifying the effects of tolerization.
Discussion
In this study, we demonstrated for the first time that exposure to MS as a model of early-life psychological stress during the induction phase of immune tolerance in childhood leads to Th2-biased sensitization and allergic airway inflammation in response to inhaled allergen in adulthood (Figs. 3, 4, 5 and 6). The prevention of the induction of immune tolerance by MS exposure was confirmed by the re-emergence of antigen-specific spleen cell proliferation and IL-4 secretion in MS/tolerized mice, both of which were suppressed in tolerized mice (Fig. 7). Furthermore, MS/tolerized mice showed lower Treg cell levels in BLNs compared to non-MS/tolerized mice (Fig. 8). Thus, stress during the tolerance-induction phase may lead to insufficient differentiation of Treg cells and subsequent failure to prevent Th2-biased immune sensitization.
Antigen exposure in early life through the mucosal route can lead to a predisposition for tolerogenic responses. In mice, the inhalation of OVA by PND 7 has been demonstrated to prevent the generation of OVA-specific IgE, to decrease the accumulation of granulocytes in the BALF, and to reduce the IL-13 levels in the BALF after sensitization and challenge with OVA in adults (Wang and McCusker 2006). In humans, maturation of innate and adaptive immunity, including immune tolerance, is essentially completed by childhood (Lloyd and Marsland 2017). Infants are particularly vulnerable to the effects of psychosocial stress on immune system development, including immune tolerance, because this system is still developing and remains highly reactive and labile for the first 2 years of life (Bosquet Enlow et al. 2014; McLaughlin et al. 2015). Indeed, exposure to psychological stress in critical developmental windows, including early childhood, can result in permanently altered changes to stress-response systems, including the autonomic, neuroendocrine system, and the immune system, which are thought to play a role in the development of respiratory disorders, including asthma (Wright 2005, 2007). Based on these findings, investigating the relationship between immune tolerance and early-life stress may help to clarify the mechanisms of asthma development in adulthood.
Previous studies have shown that neutrophils activated in vitro mediated the migration of eosinophils (Kikuchi et al. 2006; Nishihara et al. 2015). Therefore, in the current study, it is possible that the increased neutrophils in MS/tolerized mice had influenced the migration of eosinophils into the airway. IL-17A, produced mainly by Th17, has been demonstrated to play a critical role in the induction of neutrophilic airway inflammation in severe asthma (Chesne et al. 2014; Ray and Kolls 2017). We previously observed using an adult murine model of allergic asthma that restraint stress as the psychological stress induced the airway infiltration of not only eosinophils but also neutrophils (Kawano et al. 2018), and that the neutrophil accumulation was associated with the increase of IL-17A contents and Th17 numbers in the lung of the stress/tolerized mice compared with the non-stress/tolerized mice (unpublished data). Therefore, also in the MS/tolerized mice, the development of Th17 cells and the production of IL-17A by stress exposure might be involved in the infiltration of neutrophils, which would need to be investigated.
The inhibitory effects of MS exposure on Treg cell development are supported by human studies showing that traumatic and mental stress each reduce the number Treg cells in peripheral blood (Sommershof et al. 2009; Freier et al. 2010; Jergovic et al. 2014). Although the pathways linking stress exposure and Treg cell levels have not yet been determined, it has been shown that the stress hormone glucocorticoid (GC), released upon stress-triggered activation of the hypothalamic-pituitary-adrenal cortex axis, may play a critical role in stress-induced perturbation of immune tolerance. Indeed, GC is recognized worldwide as being effective for treating and preventing asthma due to its anti-allergic activities. GC’s mechanisms of action include reducing airway inflammation and decreasing airway hyper-responsiveness by limiting cytokine production in T cells and epithelial cells and thus impairing the recruitment, survival, and activation of eosinophils and other inflammatory cells (John et al. 1998; Fahy 2015). However, GC has been shown to amplify Th2 immune responses via several mechanisms. GC treatment can potentiate Th2 differentiation by either reducing a pivotal Th1-associated cytokine, IL-12, secretion from antigen-presenting cells (DeKruyff et al. 1998) or by directly suppressing Th1 cell polarization (Miyaura and Iwata 2002). Transient GC administration during sensitization was shown to permanently enhance Th2 responses in an allergic mouse model (Wiley et al. 2004). In addition, GC treatment during antigen inhalation to promote the development of respiratory tolerance was shown to eliminate IL-10-producing dendritic cells, which are required for the development of IL-10-producing Treg cells, leading to Th2-biased sensitization and allergic airway responses upon antigen exposure (Stock et al. 2005). Interestingly, the development of respiratory tolerance was shown to be prevented by systemic, but not local, GC administration (Kerzerho et al. 2012), in accordance with our finding that respiratory tolerance was inhibited in mice that showed increased blood GC levels in response to stress exposure (Fig. 2). Systemic administration of GC to immunocompetent humans and naïve mice significantly reduced the number, but not the suppressive function, of Foxp3+ Treg cells in the blood (Sbiera et al. 2011). More importantly, systemic GC administration and local GC administration were each shown to decrease the number of Foxp3+ Treg cells in mouse lungs during sensitization and allergen-induced inflammation (Olsen et al. 2015). In addition, we previously demonstrated that restraint as a psychological stressor in adulthood inhibited the development of Treg cells through the action of glucocorticoids (Kawano et al. 2018), indicated by the result that a GC-receptor antagonist administered before each stress exposure during the tolerance-induction phase eliminated the anti-tolerogenic effects of psychological stress. Previous studies showed that the transfer of transforming growth factor β (Polte and Hansen 2008; Verhasselt et al. 2008, Verhasselt 2010) or the antigen-immunoglobulin complex (Mosconi et al. 2010; Ohsaki et al. 2018) through breast milk of tolerized-mother mice was important for the induction of immune tolerance in the newborns by lactation. On the other hand, in the current study, the tolerance in pups was induced by OVA inhalation directly to pups but not to mother mice. Furthermore, MS showed no effect on the asthmatic responses under non-tolerized condition. Accordingly, the interruption of lactation might be considered to have little effect on the acquisition of tolerance in pups, although the influence of the interruption of lactation is not entirely excluded.
Recent studies have investigated the mechanisms underlying the absence of immune tolerance associated with increased susceptibility to allergic asthma. Viral respiratory infections in early life are risk factors for developing asthma in adult life. Simultaneous infection with rhinovirus and induction of tolerance in mice increased susceptibility to allergic airway responses by inhibiting the acquisition of immune tolerance induced by intranasal OVA and reducing the development of Foxp3+ Treg cells through the expression of Th2-oriented molecules such as OX40 ligand expression by lung dendritic cells and IL-33 and thymic stromal lymphopoietin expression by airway epithelial cells (Mehta et al. 2016). After OVA tolerization by breast milk from OVA-exposed maternal mice, recurrent RSV infection in pups induced allergic responses in adult life (6 weeks old), while uninfected pups showed no such responses. RSV infection was also shown to alter the phenotype of Foxp3+ Treg cells in an IL-4 dependent pathway, including GATA-3 expression, Th2 cytokine production, and loss of suppressive activity (Krishnamoorthy et al. 2012). The potential of IL-33 to overcome acquired immune tolerance was confirmed by a study that showed that administrating intranasal IL-33 to tolerized mice (during or after sensitization) caused allergen-induced airway inflammation and that IL-33 binds Treg cells and thus evokes Th2-like properties by inducing GATA-3 expression and Th2 cytokine synthesis, which have been associated with a loss of suppressive function (Chen et al. 2017). The ability of airway epithelial cells to overwhelm acquired immune tolerance was also demonstrated using another allergic murine model. NF-κB activation in airway epithelial cells during sensitization elicited allergic airway responses in tolerized mice in association with the activation of myeloid dendritic cells required for allergic responses in IL-4- and IL-1-dependent manners (Ather et al. 2015). There might be distinctive mechanisms which impair respiratory tolerance leading to increased susceptibility to allergic asthma. These mechanisms might depend on causative factors, such as activation of endocrine pathways by psychological stress, as in the current study, or inflammatory processes by viral infection (Krishnamoorthy et al. 2012; Mehta et al. 2016), or they may depend on developmental time points, such as insufficient Treg cell development during the induction phase of immune tolerance (Mehta et al. 2016) or Th2-like-phenotypic changes of Treg cells (Krishnamoorthy et al. 2012) or development of Th2-oriented dendritic cells (Huang et al. 2014) after the acquisition of immune tolerance.
In conclusion, we provide the evidence that MS as a psychological stressor inhibits the development of respiratory tolerance by suppressing Treg cell induction, resulting in increased susceptibility to allergic asthma through Th2-biased immune sensitization. Future studies are necessary to explore the neuroendocrine pathway linking stress exposure to GC release, including the role of neuropeptides involved in stress-related asthma exacerbations, such as opioids (Drolet et al. 2001; Miyasaka et al. 2018), as well as the endocrine-immune pathway, especially with regards to the interaction between GCs and dendritic cells leading to impaired immune tolerance.
Acknowledgments
This work was supported in part by a Grant-in-Aid for Scientific Research (C) (Nos. 25461164, 17K09624), a Grant-in-Aid for Young Scientists (B) (Nos. 16K19608, 17K16212), and the Matching Fund Subsidy for Private Universities (Nos. S1001002, S1511001L) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan. The funders played no role in the study design, the collection, analysis, and interpretation of data, or the preparation of the manuscript.
Conflict of Interest
The authors declare no conflict of interest.
References
-
Ather,
J.L.,
Foley,
K.L.,
Suratt,
B.T.,
Boyson,
J.E. &
Poynter,
M.E.
(2015) Airway epithelial NF-κB activation promotes the ability to overcome inhalational antigen tolerance. Clin. Exp. Allergy, 45, 1245-1258.
-
Barczyk,
A.,
Pierzchala,
W.,
Caramori,
G.,
Wiaderkiewicz,
R.,
Kaminski,
M.,
Barnes,
P.J. &
Adcock,
I.M.
(2014) Decreased percentage of CD4(+)Foxp3(+)TGF-β(+) and increased percentage of CD4(+)IL-17(+) cells in bronchoalveolar lavage of asthmatics. J. Inflamm. (Lond), 11, 22.
-
Bosquet Enlow,
M.,
King,
L.,
Schreier,
H.M.,
Howard,
J.M.,
Rosenfield,
D.,
Ritz,
T. &
Wright,
R.J.
(2014) Maternal sensitivity and infant autonomic and endocrine stress responses. Early Hum. Dev., 90, 377-385.
-
Chen,
C.C.,
Kobayashi,
T.,
Iijima,
K.,
Hsu,
F.C. &
Kita,
H.
(2017) IL-33 dysregulates regulatory T cells and impairs established immunologic tolerance in the lungs. J. Allergy Clin. Immunol., 140, 1351-1363.
-
Chesne,
J.,
Braza,
F.,
Mahay,
G.,
Brouard,
S.,
Aronica,
M. &
Magnan,
A.
(2014) IL-17 in severe asthma. Where do we stand? Am. J. Respir. Crit. Care Med., 190, 1094-1101.
-
Coogan,
P.F.,
Wise,
L.A.,
O’Connor,
G.T.,
Brown,
T.A.,
Palmer,
J.R. &
Rosenberg,
L.
(2013) Abuse during childhood and adolescence and risk of adult-onset asthma in African American women. J. Allergy Clin. Immunol., 131, 1058-1063.
-
DeKruyff,
R.H.,
Fang,
Y. &
Umetsu,
D.T.
(1998) Corticosteroids enhance the capacity of macrophages to induce Th2 cytokine synthesis in CD4+ lymphocytes by inhibiting IL-12 production. J. Immunol., 160, 2231-2237.
-
Drolet,
G.,
Dumont,
E.C.,
Gosselin,
I.,
Kinkead,
R.,
Laforest,
S. &
Trottier,
J.F.
(2001) Role of endogenous opioid system in the regulation of the stress response. Prog. Neuropsychopharmacol. Biol. Psychiatry, 25, 729-741.
-
Fahy,
J.V.
(2015) Type 2 inflammation in asthma: present in most, absent in many. Nat. Rev. Immunol., 15, 57-65.
-
Freier,
E.,
Weber,
C.S.,
Nowottne,
U.,
Horn,
C.,
Bartels,
K.,
Meyer,
S.,
Hildebrandt,
Y.,
Luetkens,
T.,
Cao,
Y.,
Pabst,
C.,
Muzzulini,
J.,
Schnee,
B.,
Brunner-Weinzierl,
M.C.,
Marangolo,
M.,
Bokemeyer,
C.,
et al. (2010) Decrease of CD4(+)FOXP3(+) T regulatory cells in the peripheral blood of human subjects undergoing a mental stressor. Psychoneuroendocrinology, 35, 663-673.
-
Fujita,
H.,
Meyer,
N.,
Akdis,
M. &
Akdis,
C.A.
(2012) Mechanisms of immune tolerance to allergens. Chem. Immunol. Allergy, 96, 30-38.
-
Hartl,
D.,
Koller,
B.,
Mehlhorn,
A.T.,
Reinhardt,
D.,
Nicolai,
T.,
Schendel,
D.J.,
Griese,
M. &
Krauss-Etschmann,
S.
(2007) Quantitative and functional impairment of pulmonary CD4+CD25hi regulatory T cells in pediatric asthma. J. Allergy Clin. Immunol., 119, 1258-1266.
-
Holgate,
S.T.,
Wenzel,
S.,
Postma,
D.S.,
Weiss,
S.T.,
Renz,
H. &
Sly,
P.D.
(2015) Asthma. Nat. Rev. Dis. Primers, 1, 15025.
-
Huang,
X.M.,
Liu,
X.S.,
Lin,
X.K.,
Yu,
H.,
Sun,
J.Y.,
Liu,
X.K.,
Chen,
C.,
Jin,
H.L.,
Zhang,
G.E.,
Shi,
X.X.,
Zhang,
Q. &
Yu,
J.R.
(2014) Role of plasmacytoid dendritic cells and inducible costimulator-positive regulatory T cells in the immunosuppression microenvironment of gastric cancer. Cancer Sci., 105, 150-158.
-
Ilmarinen,
P.,
Tuomisto,
L.E. &
Kankaanranta,
H.
(2015) Phenotypes, risk factors, and mechanisms of adult-onset asthma. Mediators Inflamm., 2015, 514868.
-
Jergovic,
M.,
Bendelja,
K.,
Vidovic,
A.,
Savic,
A.,
Vojvoda,
V.,
Aberle,
N.,
Rabatic,
S.,
Jovanovic,
T. &
Sabioncello,
A.
(2014) Patients with posttraumatic stress disorder exhibit an altered phenotype of regulatory T cells. Allergy Asthma Clin. Immunol., 10, 43.
-
John,
M.,
Lim,
S.,
Seybold,
J.,
Jose,
P.,
Robichaud,
A.,
O’Connor,
B.,
Barnes,
P.J. &
Chung,
K.F.
(1998) Inhaled corticosteroids increase interleukin-10 but reduce macrophage inflammatory protein-1alpha, granulocyte-macrophage colony-stimulating factor, and interferon-gamma release from alveolar macrophages in asthma. Am. J. Respir. Crit. Care Med., 157, 256-262.
-
Kawano,
T.,
Ouchi,
R.,
Ishigaki,
T.,
Masuda,
C.,
Miyasaka,
T.,
Ohkawara,
Y.,
Ohta,
N.,
Takayanagi,
M.,
Takahashi,
T. &
Ohno,
I.
(2018) Increased susceptibility to allergic asthma with the impairment of respiratory tolerance caused by psychological stress. Int. Arch. Allergy Immunol., 177, 1-15.
-
Kerzerho,
J.,
Wunsch,
D.,
Szely,
N.,
Meyer,
H.A.,
Lurz,
L.,
Rose,
L.,
Wahn,
U.,
Akbari,
O. &
Stock,
P.
(2012) Effects of systemic versus local administration of corticosteroids on mucosal tolerance. J. Immunol., 188, 470-476.
-
Kikuchi,
I.,
Kikuchi,
S.,
Kobayashi,
T.,
Hagiwara,
K.,
Sakamoto,
Y.,
Kanazawa,
M. &
Nagata,
M.
(2006) Eosinophil trans-basement membrane migration induced by interleukin-8 and neutrophils. Am. J. Respir. Cell Mol. Biol., 34, 760-765.
-
Kim,
R.Y.,
Rae,
B.,
Neal,
R.,
Donovan,
C.,
Pinkerton,
J.,
Balachandran,
L.,
Starkey,
M.R.,
Knight,
D.A.,
Horvat,
J.C. &
Hansbro,
P.M.
(2016) Elucidating novel disease mechanisms in severe asthma. Clin. Transl. Immunology, 5, e91.
-
Krishnamoorthy,
N.,
Khare,
A.,
Oriss,
T.B.,
Raundhal,
M.,
Morse,
C.,
Yarlagadda,
M.,
Wenzel,
S.E.,
Moore,
M.L.,
Peebles,
R.S. Jr.,
Ray,
A. &
Ray,
P.
(2012) Early infection with respiratory syncytial virus impairs regulatory T cell function and increases susceptibility to allergic asthma. Nat. Med., 18, 1525-1530.
-
Lambrecht,
B.N. &
Hammad,
H.
(2015) The immunology of asthma. Nat. Immunol., 16, 45-56.
-
Lloyd,
C.M. &
Hawrylowicz,
C.M.
(2009) Regulatory T cells in asthma. Immunity, 31, 438-449.
-
Lloyd,
C.M. &
Marsland,
B.J.
(2017) Lung homeostasis: influence of age, microbes, and the immune system. Immunity, 46, 549-561.
-
Loerbroks,
A.,
Apfelbacher,
C.J.,
Thayer,
J.F.,
Debling,
D. &
Sturmer,
T.
(2009) Neuroticism, extraversion, stressful life events and asthma: a cohort study of middle-aged adults. Allergy, 64, 1444-1450.
-
Mamessier,
E.,
Nieves,
A.,
Lorec,
A.M.,
Dupuy,
P.,
Pinot,
D.,
Pinet,
C.,
Vervloet,
D. &
Magnan,
A.
(2008) T-cell activation during exacerbations: a longitudinal study in refractory asthma. Allergy, 63, 1202-1210.
-
Maniam,
J.,
Antoniadis,
C. &
Morris,
M.J.
(2014) Early-life stress, HPA axis adaptation, and mechanisms contributing to later health outcomes. Front. Endocrinol. (Lausanne), 5, 73.
-
Martin-Orozco,
E.,
Norte-Munoz,
M. &
Martinez-Garcia,
J.
(2017) Regulatory T cells in allergy and asthma. Front. Pediatr., 5, 117.
-
McLaughlin,
K.A.,
Sheridan,
M.A.,
Tibu,
F.,
Fox,
N.A.,
Zeanah,
C.H. &
Nelson,
C.A. 3rd
(2015) Causal effects of the early caregiving environment on development of stress response systems in children. Proc. Natl. Acad. Sci. USA, 112, 5637-5642.
-
Mehta,
A.K.,
Duan,
W.,
Doerner,
A.M.,
Traves,
S.L.,
Broide,
D.H.,
Proud,
D.,
Zuraw,
B.L. &
Croft,
M.
(2016) Rhinovirus infection interferes with induction of tolerance to aeroantigens through OX40 ligand, thymic stromal lymphopoietin, and IL-33. J. Allergy Clin. Immunol., 137, 278-288.
-
Miyasaka,
T.,
Dobashi-Okuyama,
K.,
Takahashi,
T.,
Takayanagi,
M. &
Ohno,
I.
(2018) The interplay between neuroendocrine activity and psychological stress-induced exacerbation of allergic asthma. Allergol. Int., 67, 32-42.
-
Miyaura,
H. &
Iwata,
M.
(2002) Direct and indirect inhibition of Th1 development by progesterone and glucocorticoids. J. Immunol., 168, 1087-1094.
-
Mosconi,
E.,
Rekima,
A.,
Seitz-Polski,
B.,
Kanda,
A.,
Fleury,
S.,
Tissandie,
E.,
Monteiro,
R.,
Dombrowicz,
D.D.,
Julia,
V.,
Glaichenhaus,
N. &
Verhasselt,
V.
(2010) Breast milk immune complexes are potent inducers of oral tolerance in neonates and prevent asthma development. Mucosal Immunol., 3, 461-474.
-
Murgatroyd,
C. &
Spengler,
D.
(2011) Epigenetic programming of the HPA axis: early life decides. Stress, 14, 581-589.
-
Nishi,
M.,
Horii-Hayashi,
N. &
Sasagawa,
T.
(2014) Effects of early life adverse experiences on the brain: implications from maternal separation models in rodents. Front. Neurosci., 8, 166.
-
Nishihara,
F.,
Nakagome,
K.,
Kobayashi,
T.,
Noguchi,
T.,
Araki,
R.,
Uchida,
Y.,
Soma,
T. &
Nagata,
M.
(2015) Trans-basement membrane migration of eosinophils induced by LPS-stimulated neutrophils from human peripheral blood in vitro. ERJ Open Res., 1.
-
Noval Rivas,
M. &
Chatila,
T.A.
(2016) Regulatory T cells in allergic diseases. J. Allergy Clin. Immunol., 138, 639-652.
-
Ohsaki,
A.,
Venturelli,
N.,
Buccigrosso,
T.M.,
Osganian,
S.K.,
Lee,
J.,
Blumberg,
R.S. &
Oyoshi,
M.K.
(2018) Maternal IgG immune complexes induce food allergen-specific tolerance in offspring. J. Exp. Med., 215, 91-113.
-
Olsen,
P.C.,
Kitoko,
J.Z.,
Ferreira,
T.P.,
de-Azevedo,
C.T.,
Arantes,
A.C. &
Martins Mu,
A.
(2015) Glucocorticoids decrease Treg cell numbers in lungs of allergic mice. Eur. J. Pharmacol., 747, 52-58.
-
Palomares,
O.,
Martin-Fontecha,
M.,
Lauener,
R.,
Traidl-Hoffmann,
C.,
Cavkaytar,
O.,
Akdis,
M. &
Akdis,
C.A.
(2014) Regulatory T cells and immune regulation of allergic diseases: roles of IL-10 and TGF-β. Genes Immun., 15, 511-520.
-
Palomares,
O.,
Yaman,
G.,
Azkur,
A.K.,
Akkoc,
T.,
Akdis,
M. &
Akdis,
C.A.
(2010) Role of Treg in immune regulation of allergic diseases. Eur. J. Immunol., 40, 1232-1240.
-
Peterson,
R.A.
(2012) Regulatory T-cells: diverse phenotypes integral to immune homeostasis and suppression. Toxicol. Pathol., 40, 186-204.
-
Polte,
T. &
Hansen,
G.
(2008) Maternal tolerance achieved during pregnancy is transferred to the offspring via breast milk and persistently protects the offspring from allergic asthma. Clin. Exp. Allergy, 38, 1950-1958.
-
Ray,
A. &
Kolls,
J.K.
(2017) Neutrophilic Inflammation in Asthma and Association with Disease Severity. Trends Immunol., 38, 942-954.
-
Rosa,
M.J.,
Lee,
A.G. &
Wright,
R.J.
(2018) Evidence establishing a link between prenatal and early-life stress and asthma development. Curr. Opin. Allergy Clin. Immunol., 18, 148-158.
-
Rothenberg,
M.E.,
Saito,
H. &
Peebles,
R.S. Jr.
(2017) Advances in mechanisms of allergic disease in 2016. J. Allergy Clin. Immunol., 140, 1622-1631.
-
Sakaguchi,
S.,
Miyara,
M.,
Costantino,
C.M. &
Hafler,
D.A.
(2010) FOXP3+ regulatory T cells in the human immune system. Nat. Rev. Immunol., 10, 490-500.
-
Sbiera,
S.,
Dexneit,
T.,
Reichardt,
S.D.,
Michel,
K.D.,
van den Brandt,
J.,
Schmull,
S.,
Kraus,
L.,
Beyer,
M.,
Mlynski,
R.,
Wortmann,
S.,
Allolio,
B.,
Reichardt,
H.M. &
Fassnacht,
M.
(2011) Influence of short-term glucocorticoid therapy on regulatory T cells in vivo. PLoS One, 6, e24345.
-
Scott,
K.M.,
Smith,
D.A. &
Ellis,
P.M.
(2012) A population study of childhood maltreatment and asthma diagnosis: differential associations between child protection database versus retrospective self-reported data. Psychosom. Med., 74, 817-823.
-
Sommershof,
A.,
Aichinger,
H.,
Engler,
H.,
Adenauer,
H.,
Catani,
C.,
Boneberg,
E.M.,
Elbert,
T.,
Groettrup,
M. &
Kolassa,
I.T.
(2009) Substantial reduction of naive and regulatory T cells following traumatic stress. Brain Behav. Immun., 23, 1117-1124.
-
Stock,
P.,
Akbari,
O.,
DeKruyff,
R.H. &
Umetsu,
D.T.
(2005) Respiratory tolerance is inhibited by the administration of corticosteroids. J. Immunol., 175, 7380-7387.
-
Tarullo,
A.R. &
Gunnar,
M.R.
(2006) Child maltreatment and the developing HPA axis. Horm. Behav., 50, 632-639.
-
To,
T.,
Stanojevic,
S.,
Moores,
G.,
Gershon,
A.S.,
Bateman,
E.D.,
Cruz,
A.A. &
Boulet,
L.P.
(2012) Global asthma prevalence in adults: findings from the cross-sectional world health survey. BMC Public Health, 12, 204.
-
Tractenberg,
S.G.,
Levandowski,
M.L.,
de Azeredo,
L.A.,
Orso,
R.,
Roithmann,
L.G.,
Hoffmann,
E.S.,
Brenhouse,
H. &
Grassi-Oliveira,
R.
(2016) An overview of maternal separation effects on behavioural outcomes in mice: evidence from a four-stage methodological systematic review. Neurosci. Biobehav. Rev., 68, 489-503.
-
Verhasselt,
V.
(2010) Neonatal tolerance under breastfeeding influence: the presence of allergen and transforming growth factor-beta in breast milk protects the progeny from allergic asthma. J. Pediatr., 156, S16-20.
-
Verhasselt,
V.,
Milcent,
V.,
Cazareth,
J.,
Kanda,
A.,
Fleury,
S.,
Dombrowicz,
D.,
Glaichenhaus,
N. &
Julia,
V.
(2008) Breast milk-mediated transfer of an antigen induces tolerance and protection from allergic asthma. Nat. Med., 14, 170-175.
-
Wang,
Y. &
McCusker,
C.
(2006) Neonatal exposure with LPS and/or allergen prevents experimental allergic airways disease: development of tolerance using environmental antigens. J. Allergy Clin. Immunol., 118, 143-151.
-
Wiley,
R.E.,
Cwiartka,
M.,
Alvarez,
D.,
Mackenzie,
D.C.,
Johnson,
J.R.,
Goncharova,
S.,
Lundblad,
L. &
Jordana,
M.
(2004) Transient corticosteroid treatment permanently amplifies the Th2 response in a murine model of asthma. J. Immunol., 172, 4995-5005.
-
Wright,
R.J.
(2005) Stress and atopic disorders. J. Allergy Clin. Immunol., 116, 1301-1306.
-
Wright,
R.J.
(2007) Prenatal maternal stress and early caregiving experiences: implications for childhood asthma risk. Paediatr. Perinat. Epidemiol., 21 Suppl. 3, 8-14.
-
Zhou,
W.,
Goleniewska,
K.,
Zhang,
J.,
Dulek,
D.E.,
Toki,
S.,
Lotz,
M.T.,
Newcomb,
D.C.,
Boswell,
M.G.,
Polosukhin,
V.V.,
Milne,
G.L.,
Wu,
P.,
Moore,
M.L.,
FitzGerald,
G.A. &
Peebles,
R.S. Jr.
(2014) Cyclooxygenase inhibition abrogates aeroallergen-induced immune tolerance by suppressing prostaglandin I2 receptor signaling. J. Allergy Clin. Immunol., 134, 698-705.



