Abstract
Lung cancer represents the leading cause of cancer-associated mortality, and non-small-cell lung cancer (NSCLC) is the most frequent histologic sub-type. It is therefore urgent to develop novel agents for the treatment of NSCLC. Fraxetin (FXT) is a potent plant-derived product and has been recognized as a promising anticancer agent for breast cancer and osteosarcoma. However, the anti-cancer potential of FXT for NSCLC remains to be elucidated. Accordingly, in the present study, we evaluated the inhibitory effect of FXT on the proliferation and growth of NSCLC cells using six human NSCLC cell lines: A549, H460, HCC827, H1650, PC-9 and H1975. FXT exhibited significant inhibitory effects on the proliferation of these cancer cell lines. By contrast, no inhibitory effect was observed on the viability of non-cancer lung cell lines even at the highest concentration of FXT (100 μM). Among the NSCLC cell lines, HCC827 and H1650 cells showed the most sensitive to FXT. Accordingly, HCC827 and H1650 cells were used for the subsequent experiments. Flow cytometric analysis revealed that FXT caused a significant cell cycle arrest and pro-apoptotic effects. Mechanistically, FXT suppressed the IL-6-induced phosphorylation of tyrosine residue (Tyr705) of signal transducer and activator of transcription 3 (STAT3) probably by binding to STAT3. Molecular docking and molecular dynamic simulations studies indicated that FXT interacts with STAT3 through hydrogen bond and hydrophobic interaction. In conclusion, these findings suggest that FXT could be a promising lead compound to be used as a novel STAT3 inhibitor and potential antitumor agent for the treatment of NSCLC.
Introduction
Lung cancer represents the leading cause of cancer-associated mortality accounting for an estimated 1.76 million deaths worldwide in 2018 worldwide (Bray et al. 2018). Non-small-cell lung cancer (NSCLC) is the most frequent histologic sub-type, accounting for approximately 80%-85% of all cases of lung cancer, for which the predicted 5-year survival rate is extremely poor (Reck et al. 2013). Although numerous novel targeted therapies have brought significant improvements to the survival and prognosis for patients with NSCLC, the overall outcome of current therapies for NSCLC remain obscure (Hirsch et al. 2017). Besides, the standard first-line treatment for patients with advanced NSCLC is platinum-based doublet chemotherapy (Rossi and Di Maio 2016); however, these agents are commonly associated with nephrotoxicity, neurotoxicity, and myelosuppression. Thus, it becomes imperative to identify novel agents with promising antitumor activity, less toxicity, and high therapeutic index for effective treatment of patients with advanced NSCLC.
Coumarin (2H-1-benzopyran-2-one), the parent molecule of coumarin derivatives, belongs to the family of the benzopyrone consisting of benzene ring joined to the α-pyrone nucleus and is comprised of a large class of naturally occurring phenolic substances found in plants. Coumarin and its derivatives are identified to exhibit diverse pharmacological activities including anticoagulant, anti-inflammatory, anti-microbial and particularly anti-tumor activities, antioxidant, and neuroprotective properties (Peng et al. 2013; Emami and Dadashpour 2015; Pinto and Silva 2017). In recent years, coumarin-based compounds have become a hotspot for anticancer drug design and discovery (Kaur et al. 2015). Natural products, esculetin and osthole, are the two well-studied coumarin-based anticancer agents that exhibit potent in vitro cytotoxic activity against the growth of several human tumor cell lines (gastric carcinoma, colon carcinoma, and breast cancer cell line) and in vivo anti-tumor activities in xenograft models (Lee et al. 2013; Wang et al. 2017; Dai et al. 2018; Xu et al. 2018). Besides, several natural and synthetic coumarin-based drugs, including Warfarin (anti-metastasis agent) and Geiparvarin (anti-proliferation agent), have been screened and found exhibit specific anticancer effects, suggesting that these cytotoxic coumarin-based compounds represent a desirable lead for novel anticancer drug discovery with minimized toxicity to normal tissues (Chimichi et al. 2009; Kirane et al. 2015).
Fraxetin (FXT, 7,8-dihydroxy-6-methoxy coumarin) is a plant-derived coumarin primarily isolated Fraxinus bungeana and has been reported to exert potent antibacterial, anti-inflammatory, and neuroprotective effects (Molina-Jimenez et al. 2004; Wang et al. 2014; Chen et al. 2018). Recently, studies have reported that FXT could induce apoptosis and suppress metastasis in certain tumors. Liu et al. (2017) identified that FXT significantly facilitated the apoptosis in MCF-7 breast cancer cells through upregulation the expression of Fas, FasL and Bax. Moreover, Kimura and Sumiyoshi (2015) demonstrated that FXT inhibits the proliferation of osteosarcoma LM8 cells in vitro and the tumor growth in mice implanted with LM8 cells in vivo. However, there is a paucity of studies on the anti-proliferative activities of FXT. Thus, the anti-cancer potential of FXT remains to be elucidated.
In the present study, we investigated the in vitro potential of FXT in a panel of six NSCLC cell lines. The results from cell proliferation (MTS assay) and colony formation experiments indicated that FXT significantly inhibited the proliferation and growth of NSCLC cells. Flow cytometric and Western blot analyses indicated that FXT significantly effectively arrested NSCLC cells at the G0/G1phase and induced apoptosis. The further mechanistic investigation revealed FXT as a potential inhibitor of signal transducer and activator of transcription 3 (STAT3). Eventually, the possible mechanism on the interaction between STAT3 and FXT was simulated through molecular docking and molecular simulations, which demonstrated that the hydrogen bond and hydrophobic interaction might contribute to their binding interaction.
Material and Methods
Cell culture and reagents
The human pulmonary epithelial cells Beas-2B, human bronchial epithelial cells HBE and NSCLC cell lines A549, H460, HCC827, H1650, PC-9 and H1975 were obtained from Shanghai Institute of Biosciences and Cell Resources Center (Chinese Academy of Sciences, Shanghai, China). The cells were routinely cultured in RPMI-1640 or DMEM (Gibco/BRL lifeTechnologies, Eggenstein, Germany) supplemented with 10% fetal bovine serum FBS (Hyclone, Logan, UT), 1% penicillin/streptomycin solution in a humidified atmosphere of 5% CO2 at 37°C. 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) powder was purchased from Promega (Wisconsin, USA). The primary antibodies used in this study, including P-STAT3 (Y705), STAT3, Bax, Bcl-2, Cleaved PARP, Cyclin D1, Cdk-4, Cdk-6 and β-actin were all purchased from Cell Signaling Technology (Danvers, MA). Goat anti-rabbit IgG-HRP secondary antibody were obtained from Santa Cruz Biotechnology (Santa Cruz, CA); FXT was purchased from Solorbio (Beijing, China).
Cell viability assay
The cell viability was measured by the Cell Titer 96 Aqueous Non-Radioactive Cell Proliferation Assay Kit (Promega). 5×103 cells were seeded in 96-well plates overnight, then the culture medium was removed and all cells were treated with different concentrations of chemicals for 72 h. The absorbance value of each well was determined by a microplate reader at 490 nm. Each experiment was repeated for three times.
Colony formation assay
HCC827 and H1650 cells (1,000 cells/well) were seeded in 6-well cell culture plate for 24 h, then DMSO and FXT (20 or 40 µM) were added to the culture medium. After 24 h, the culture medium was replaced with fresh culture medium for 14 days. Colonies was removed the medium, washed with PBS twice, fixed with methanol for 15 min, then washed with PBS three times, and finally stained with crystal violet for 15 min. Colonies containing more than 50 cells were counted, and visualized colonies were then photographed.
Cell apoptosis analysis
Cell apoptosis was detected by flow cytometer analysis and Western blotting. For flow cytometer analysis of apoptosis, cells treated as indicated for 24 h were harvested by trypsin, washed twice by PBS, and re-suspended in 100 μL 1× binding buffer. 5 μL FITC Annexin V and PI (556547, BD Biosciences, USA) was added to the cell suspension and then incubated for 15 min at room temperature. After dilution with 400 μL binding buffer, the samples were analyzed by ACS Calibur flow cytometer (BD). For apoptosis by Western blotting, cleavage of PARP (poly ADP-ribose polymerase), Bcl-2 and Bax were analyzed.
Cell cycle analysis
Cells (3 × 105 cells/well) were seeded in 6-well plates and allowed to adhere overnight. The next day, the cells were treated with different compounds as indicated for 24 h. Then the cells were trypsinized, washed, and fixed in 75% ice-cold ethanol at 4°C overnight. After centrifugation, the pellets were washed with cold PBS, suspended in 500 μL PBS with 50 mg/ml propidium iodide (PI) and incubated at 4°C for 30 min in the dark. Then cell suspension was subjected to a FACS Calibur instrument (Becton Dickinson FACSCalibor, BD Biosciences, Franklin Lakes, NJ).
Western blot analysis
After treated as indicated for 24 h, the cells were washed once by PBS. The cell lysates were quantitated by BCA protein assay kit (Bio-Rad Laboratories, Hercules, CA, USA). Equal amounts of proteins were separated by SDS-PAGE and transferred to PVDF membrane. After being blocked with 5% non-fat dry milk in TBST for 1.5 h, membranes were incubated with a 1:1,000 dilution of specific primary antibody overnight at 4°C and the secondary antibody conjugated with horseradish peroxidase (HRP) (1:5,000, Santa Cruz, CA) for 2 h, the immunoreactive bands were visualized with enhanced chemiluminescence (EMD Millipore, Billerica, MA, USA) in Amersham Imager 600 system (GE Healthcare Life Sciences, Shanghai, China).
Surface Plasmon Resonance (SPR) Analysis
SPR experiments were performed on a ProteOn XPR36 Protein Interaction Array system (Bio-Rad Laboratories, Hercules, CA, USA). Briefly, STAT3 solution in PBST (5 mM, pH 7.4) at a concentration of 1 mg/mL was diluted to 30 μg/mL with sodium acetate buffer (pH 4.5). The chip was activated with EDC/NHS (10 μL/min for 600 s). Then, STAT3 was loaded (5 μL/min for 400 s) and immobilized covalently. Approximately 6,000 RU of STAT3 was immobilized on the chip. Any excess of unbound STAT3 was removed by flowing PBS solution (5 mM, pH 7.4, with 5%, w/v, DMSO). FXT was prepared as 20-100 μM solution in PBS solution (5 mM, pH 7.4, with 5%, w/v, DMSO), and injected (10 μL/min for 100 s). Five concentrations were injected simultaneously at a flow rate of 30 μM/min for 120 s of association phase, followed with 120 s of dissociation phase at 25°C. The final graph was obtained by subtracting blank sensorgrams from the duplex or quadruplex sensorgrams. Data were analyzed by ProteOn manager software.
Prediction of the interaction between STAT3 and fraxetin
The STAT3 was retrieved from the Protein Data Bank (PDB) webserver (PDB code: 1BG1) (Becker et al. 1998). The structure of SH2 domain of STAT3 was selected as the binding site according to previous studies (Li et al. 2011; Xiao et al. 2015). The structure of SH2 domain was refined by PyMol, an open-source software, contained removing all water molecules, DNA, non-bonded hetero-atoms. Then, the STAT3 SH2 domain and fraxetin were processed by AutoDockTools 1.5.6 software (Morris et al. 2009). The grid box (25 Å × 25 Å × 25 Å) was covered the active binding site of STAT3. The binding mode was predicted by the AutoDock (version 4.2) software (Morris et al. 2009). Lamarckian genetic algorithm (LGA) was performed for conformational sampling with trials of 100 dockings and other settings were set as default. The conformation with lowest binding energy was chosen for further molecular dynamics (MD) simulation analysis.
Molecular dynamics (MD) simulation
The constructed structure of STAT3/fraxetin complex provided by previously was used for the MD simulations. The restrained electrostatic potential (RESP) method were used to calculate the partial atomic charges for fraxetin based on HF/6-13G* basis set. The ff14SB force field and general Amber force field (GAFF2) was assigned to STAT3 and fraxetin, respectively (Wang et al. 2004; Maier et al. 2015). The complex was then placed into a TIP3P water box. Prior to MD simulation, equilibration protocol was carried out, including minimization, heating and equilibration. Minimization procedure involved 6,000 steepest descent minimization cycles, followed by 6,000 conjugate gradient minimization cycles. After the initial minimization, the system was heated up from 0 to 300 K in 200 ps. Subsequently, unconstrained equilibration at 300 K were carried out to equilibrate the system at periodic boundary condition. Finally, 30 ns production MD simulation was conducted in the NPT ensemble. During the productive MD simulation, Temperature was controlled using Langevin dynamics while pressure was controlled using Berendsen barostat (Berendsen et al. 1984; Loncharich et al. 1992). Periodic boundary conditions were employed a 10.0 Å cutoff was used for nonbonded interactions (Essmann et al. 1995). All bonds involving hydrogen atoms were constrained using the SHAKE algorithm (Krautler et al. 2001). The coordinates were recorded every 2 ps for further numerical analysis (Roe and Cheatham 2013). Binding energy decomposition were calculated using molecular mechanics/generalized Born surface area (MM/GBSA) method with GB model of 2 (igb = 2) (Miller et al. 2012). In this study, 500 snapshots extracted from the last 20 ns MD trajectory were applied to the binding free energy decomposition as described previously (Liu et al. 2015; Xue et al. 2018).
Statistical analysis
The results are presented as the mean ± standard error (SEMs). The statistics were performed using one-way ANOVA in GraphPad Pro (GraphPad, San Diego, CA, USA). P values < 0.05 were considered statistically significant. All the experiments were repeated a minimum of three times. All of the aforementioned experiments were repeated thrice.
Results
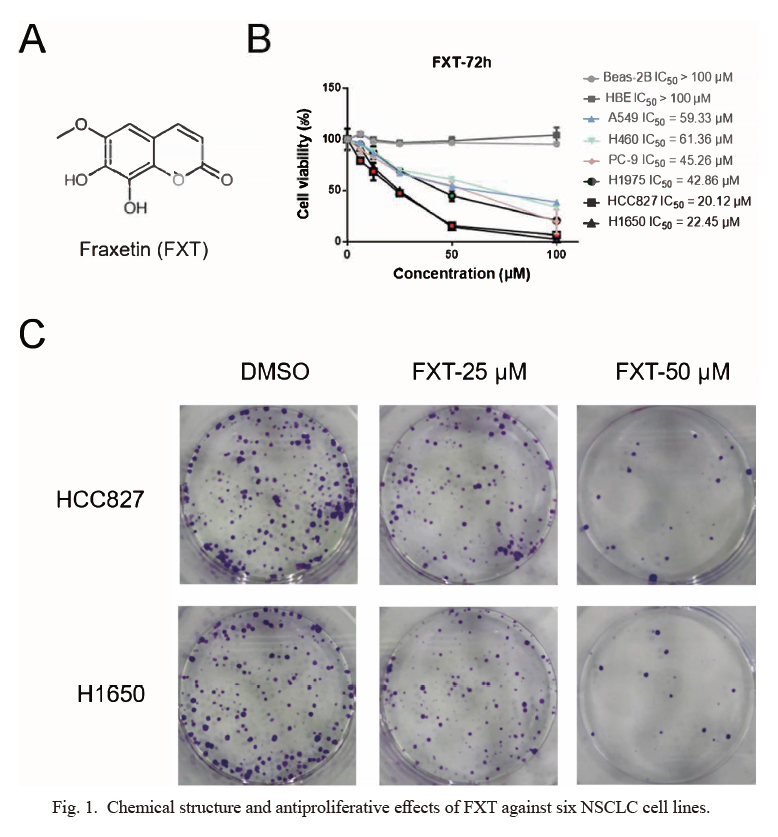
FXT inhibits NSCLC cell proliferation and colony formation
Abnormal proliferation driven by a succession of oncogenic signals is one hallmark of cancer cells. Formazan-forming assays are commonly used in cell proliferation assays. Through the reduction of tetrazolium salts by dehydrogenases and reductases to form the colorimetric formazan products, it could reflect the ratio of living cells. Using colorimetric MTS assays, the anti-proliferative effects of FXT against two non-cancer human cell lines and a panel of six NSCLC cell lines was determined (Fig. 1A). As shown in Fig. 1B, FXT exhibited significant inhibitory effects on HCC827 and H1650 cells with IC50 values of 20.12 μM and 22.45 μM, respectively. By contrast, no inhibitory effect was observed in the cell viability of non-cancer lung cell lines (Beas-2B and HBE) even at a high concentration of 100 μM. Accordingly, HCC827 and H1650 cells were used for the subsequent experiments.
Next, colony-forming assays were carried out to evaluate the long-term suppressive effects of treatment with FXT. After incubation with FXT for 2 weeks, cell colonies were stained with crystal violet and counted. Results (Fig. 1C) indicated that FXT exerted strong inhibitory effects on the clonal growth of NSCLC cells.
FXT induces NSCLC cell cycle arrest
The eukaryotic cell, as well as cancer cell, consists of four distinct phases: G1 phase, S phase, G2 phase, and M phase. A complete and ordered cell cycle is essential to cancer cell growth and survival, while arresting cells at the specific phase will severely hamper tumor progress. To determine whether FXT influences NSCLC cell cycle distribution, we analyzed the proportion of cells at different cell cycle phases by flow cytometry. The results indicated that treatment with FXT led to the accumulation of HCC827 and H1650 cells at G0/G1 phase in a dose-dependent manner (Fig. 2A). Cyclin D1 could form a complex with cdk4 and cdk6, which regulates the G1/S phase transition and may contribute to tumorigenesis. On the other hand, the results of the Western blot analysis suggested that FXT significantly down-regulated the expression of G1/S transition regulatory proteins (cyclin D1, cdk4 and cdk6, Fig. 2B). These results demonstrated that FXT effectively induced cell cycle arrest at G0/G1 phase through the reduction of the expression of cell cycle-related proteins in HCC827 and H1650 cells.

Inducing apoptosis is one of crucial means of most anticancer drugs. Caspase 3 and its substrate poly ADP-ribose polymerase (PARP) plays the central role in the transduction of programmed cell death signals. Besides, the Bcl-2 family members, such as Bcl-2 and Bax, also crucially regulate the apoptosis of cancer cells. Thus, the apoptosis-promoting effect of FXT on NSCLC cells was evaluated by double-staining with annexin V-FITC and PI. Flow cytometric analysis demonstrated that compared with the control group, early- and late-stage apoptosis rates in HCC827 and H1650 cells were both markedly increased after treatment with FXT for 24 h (Fig. 3A).
To establish molecular the mechanisms of FXT-induced apoptosis, we examined the expression levels of the apoptosis-related proteins including cleaved caspase-PARP, Bax and Bcl-2 by Western blot analysis. Fig. 3B revealed a significant upregulation of the expression of pro-apoptotic molecules cleaved-PARP and Bax, while the expression of anti-apoptotic protein bcl-2 was significantly downregulated.
FXT binds directly to STAT3 and suppresses STAT3 activation in NSCLC cells
The transcription factor STAT3 is involved in the oncogenesis of a variety of solid tumor malignancies (e.g., NSCLC, gastric cancer, breast tumors, and so on). Previously, Kimura and Sumiyoshi (2015) demonstrated that FXT suppressed the activation of IL-4 and IL-13-induced STAT3 activation in a dose-dependent manner. We therefore speculated that the antitumor activity of FXT in lung cancer might also be attributed to the inhibition of STAT3 signaling pathway. Accordingly, phosphorylation and total protein expression levels of STAT3 were analyzed by Western blot analysis. The results showed that FXT significantly decreased STAT3 phosphorylation levels in a dose-dependently manner (Fig. 4A). L-6 is a well-known tumor-promoting cytokine, as well as the significant STAT3 activating stimuli that persistently phosphorylate tyrosine residue (Tyr705) of STAT3 in most malignancies. We then determined whether FXT could inhibit IL-6-mediated phosphorylation of STAT3. Indeed, FXT inhibited the IL-6-induced phosphorylation level of STAT3 induced in a dose-dependent manner (Fig. 4B).
To investigate whether FXT interacts directly with STAT3, we performed SPR experiments. As shown in Fig. 4C, the SPR response value was increased gradually with higher concentrations of FXT and a moderate equilibrium dissociation constant (KD) of 25.65 μM. Together, these results indicate that FXT interacts with STAT3, and subsequently preventing the phosphorylation of STAT3.
Analysis of the FXT-binding site by molecular modeling
Accumulating evidence demonstrated that the STAT3 signal pathway was activated upon the phosphorylation of Tyr-705, followed by dimerization, nuclear translocation, and DNA binding. The most druggable binding pocket of the STAT3 is on the SH2 domain (Li et al. 2011). Accordingly, molecular docking was performed for prediction of the possible interaction between the SH2 domain of STAT3 and FXT. Subsequently, we performed MD simulations (30 ns) to obtain the equilibrated structures. As illustrated in Fig. 5A, the root-mean square deviations (RMSDs) of all the backbone atoms of the STAT3 SH2 domain, and the heavy atoms of FXT were analyzed to validate the stability of the studied systems. The RMSDs of the backbone atoms of the STAT3 SH2 domain exhibited a small fluctuation after 10 ns MD simulation and the heavy atoms of FXT showed relative stability during the whole simulation. These results indicate that a relatively reasonable stable conformation was obtained. After that, the last 20 ns from MD simulation trajectories were used for decomposition of the binding free energy into contribution of each residue to highlight the roles of each residue in determining protein-ligand recognition patterns. As presented in Fig. 5B, the most contributed residues were Glu-594, Arg-595, Ser-636, Ile-634 and Lys-591. Further, the detailed structural analysis indicated that FXT binds to the hydrophobic binding pocket of STAT3 to disrupt STAT3-STAT3 dimerization (Fig. 5C). The predominant interactions were hydrogen bond with Glu-594, Arg-595, Ser-636 and hydrophobic interaction with Ile-634 and Lys-591 residues (Fig. 5D).

Discussion
Natural products represent an enormous resource of bioactive compounds with abundant structural diversity and have been extensively investigated for the discovery of potential new anti-cancer agents. As an essential class of natural chemicals, coumarins were reported to exhibit immense anticancer potential with minimum side effects. Esculetin, a coumarin derivative, is recognized to exhibit anti-proliferative and pro-apoptotic effects on multiple tumor types including human colorectal carcinoma, gastric carcinoma, pancreatic cancer, and prostate cancer (Arora et al. 2016; Wang et al. 2017; Kim et al. 2018). Results from mechanistic studies also suggested that esculetin exerts its antitumor effects through the involvement of a large number of cellular pathways, including Wnt/β-catenin, Axin2/E-cadherin, and IGF-1/PI3K/Akt signaling cascades (Lee et al. 2013; Wang et al. 2017; Kim et al. 2018). Moreover, osthole, another well-known coumarin-derivative, exerts significant tumor-suppressing activities. In a recent study, Dai et al. (2018) demonstrated that osthole inhibits the proliferation of triple negative breast cancer cells both in vitro and in vivo through suppression of STAT3 activation.
In the present study, for the first time we evaluated the inhibitory effects of FXT against NSCLC cells. The findings of this study indicated that FXT significantly suppressed the proliferation and growth of NSCLC cells. By contrast, it exhibited no cytotoxicity to human pulmonary epithelial (Beas-2B) cells and human bronchial epithelial (HBE) cells. Moreover, FXT caused significant cell cycle arrest and apoptotic cell death in NSCLC cells in a dose-dependent manner. Taken together, the findings of this study suggested that FXT could be a safe and effective chemotherapeutic agent against NSCLC.
Janus kinase-STAT3 signaling axis is one of the critical pathways involved in the progression and development of various solid malignancies. Typically, STAT3 is localized in the cytoplasm and activated through phosphorylation of Tyr-705 by receptor tyrosine kinases including JAKs, EGFR, PDGFR, Src and Abl. Activated STAT3 form homo- or hetero-dimers in the cytoplasm and subsequently translocate to the nucleus to regulate the expression of target genes including cyclin D1, Myc, MMP-2, MMP-9, VEGF, and HIF1-α involved in cell proliferation, cell invasion and angiogenesis. Aberrant activation of STAT3 signaling cascade has been associated with the oncogenic potential in several malignancies (Chai et al. 2016). Furthermore, over 50% of NSCLC primary tumors and cell lines exhibit high levels of constitutively activated STAT3, suggesting that STAT3 could be a highly promising target for the treatment of lung cancer either as a single agent or in combination therapy. Several naturally-derived STAT3 inhibitors, such as curcumin, betulinic acid, and caffeic acid, were previously identified and evaluated for their anti-cancer effects in preclinical research (Jung et al. 2007; Shin et al. 2011; Xu and Zhu 2017). However, to the best of our knowledge, there are no reports on coumarin-based STAT3 inhibitor. In the present study, by Western blot analysis, we showed that FXT could restrain the activation of STAT3 in NSCLC cells. SPR analysis further confirmed that the direct interaction of FXT as a STAT3 inhibitor. Further, molecular docking and simulation analyses suggested that STAT3 inhibition through FXT predominantly depended on hydrogen bond and hydrophobic interactions with the hydrophobic cavity of the STAT3 SH2 domain. Based on the findings of modeling studies, several beneficial modification strategies were drawn. Due to the simple chemical structure of FXT, it only occupied half of the space of STAT3 SH2 hydrophobic pocket. Introduction of sizeable steric hindrance groups into the 7- or 8-hydroxy position of FXT might facilitate its binding ability. Furthermore, the rigid large conjugated structure of FXT resulted in its poor solubility. However, the introduction of a hydrophilic moiety at C-4 and C-5 position that is oriented away from the hydrophobic pocket may efficiently enhance its water solubility, which might importantly improve the poor druggability of FXT.
In summary, FXT could significantly suppress NSCLC cell proliferation, arrest the cell cycle, and induce apoptotic cell death of NSCLC cells. Mechanistically, FXT could bind directly to STAT3 and abrogate the phosphorylation of STAT3. Furthermore, molecular docking and simulations of the FXT/STAT3 complex revealed the underlying molecular mechanism of FXT through the formation of hydrogen bond and hydrophobic interaction with STAT3 SH2 domain. Overall, these data suggest that FXT may serve as a novel effective STAT3 inhibitor, and a promising candidate therapeutic agent for the treatment of NSCLC.
Acknowledgments
This work was supported by grants from the National Nature Science Foundation of China (No. 81560462) and the Applied & Basic Research Funds of Yunnan Province, China (No. 2016FB150).
Conflicts of Interest
The authors declare no conflict of interest.
References
-
Arora,
R.,
Sawney,
S.,
Saini,
V.,
Steffi,
C.,
Tiwari,
M. &
Saluja,
D.
(2016) Esculetin induces antiproliferative and apoptotic response in pancreatic cancer cells by directly binding to KEAP1. Mol. Cancer, 15, 64.
-
Becker,
S.,
Groner,
B. &
Muller,
C.W.
(1998) Three-dimensional structure of the Stat3beta homodimer bound to DNA. Nature, 394, 145-151.
-
Berendsen,
H.J.C.,
Postma,
J.P.M.,
Vangunsteren,
W.F.,
Dinola,
A. &
Haak,
J.R.
(1984) Molecular dynamics with coupling to an external bath. J. Chem. Phys., 81, 3684-3690.
-
Bray,
F.,
Ferlay,
J.,
Soerjomataram,
I.,
Siegel,
R.L.,
Torre,
L.A. &
Jemal,
A.
(2018) Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin., 68, 394-424.
-
Chai,
E.Z.,
Shanmugam,
M.K.,
Arfuso,
F.,
Dharmarajan,
A.,
Wang,
C.,
Kumar,
A.P.,
Samy,
R.P.,
Lim,
L.H.,
Wang,
L.,
Goh,
B.C.,
Ahn,
K.S.,
Hui,
K.M. &
Sethi,
G.
(2016) Targeting transcription factor STAT3 for cancer prevention and therapy. Pharmacol. Ther., 162, 86-97.
-
Chen,
X.,
Ying,
X.,
Sun,
W.,
Zhu,
H.,
Jiang,
X. &
Chen,
B.
(2018) The therapeutic effect of fraxetin on ethanol-induced hepatic fibrosis by enhancing ethanol metabolism, inhibiting oxidative stress and modulating inflammatory mediators in rats. Int. Immunopharmacol., 56, 98-104.
-
Chimichi,
S.,
Boccalini,
M.,
Salvador,
A.,
Dall’Acqua,
F.,
Basso,
G. &
Viola,
G.
(2009) Synthesis and biological evaluation of new geiparvarin derivatives. Chem. Med. Chem., 4, 769-779.
-
Dai,
X.,
Yin,
C.,
Zhang,
Y.,
Guo,
G.,
Zhao,
C.,
Wang,
O.,
Xiang,
Y.,
Zhang,
X. &
Liang,
G.
(2018) Osthole inhibits triple negative breast cancer cells by suppressing STAT3. J. Exp. Clin. Cancer Res., 37, 322.
-
Emami,
S. &
Dadashpour,
S.
(2015) Current developments of coumarin-based anti-cancer agents in medicinal chemistry. Eur. J. Med. Chem., 102, 611-630.
-
Essmann,
U.,
Perera,
L.,
Berkowitz,
M.L.,
Darden,
T.,
Lee,
H. &
Pedersen,
L.G.
(1995) A smooth particle mesh Ewald method. J. Chem. Phys., 103, 8577-8593.
-
Hirsch,
F.R.,
Scagliotti,
G.V.,
Mulshine,
J.L.,
Kwon,
R.,
Curran,
W.J. Jr.,
Wu,
Y.L. &
Paz-Ares,
L.
(2017) Lung cancer: current therapies and new targeted treatments. Lancet, 389, 299-311.
-
Jung,
J.E.,
Kim,
H.S.,
Lee,
C.S.,
Park,
D.H.,
Kim,
Y.N.,
Lee,
M.J.,
Lee,
J.W.,
Park,
J.W.,
Kim,
M.S.,
Ye,
S.K. &
Chung,
M.H.
(2007) Caffeic acid and its synthetic derivative CADPE suppress tumor angiogenesis by blocking STAT3-mediated VEGF expression in human renal carcinoma cells. Carcinogenesis, 28, 1780-1787.
-
Kaur,
M.,
Kohli,
S.,
Sandhu,
S.,
Bansal,
Y. &
Bansal,
G.
(2015) Coumarin: a promising scaffold for anticancer agents. Anticancer Agents Med. Chem., 15, 1032-1048.
-
Kim,
W.K.,
Byun,
W.S.,
Chung,
H.J.,
Oh,
J.,
Park,
H.J.,
Choi,
J.S. &
Lee,
S.K.
(2018) Esculetin suppresses tumor growth and metastasis by targeting Axin2/E-cadherin axis in colorectal cancer. Biochem. Pharmacol., 152, 71-83.
-
Kimura,
Y. &
Sumiyoshi,
M.
(2015) Antitumor and antimetastatic actions of dihydroxycoumarins (esculetin or fraxetin) through the inhibition of M2 macrophage differentiation in tumor-associated macrophages and/or G1 arrest in tumor cells. Eur. J. Pharmacol., 746, 115-125.
-
Kirane,
A.,
Ludwig,
K.F.,
Sorrelle,
N.,
Haaland,
G.,
Sandal,
T.,
Ranaweera,
R.,
Toombs,
J.E.,
Wang,
M.,
Dineen,
S.P.,
Micklem,
D.,
Dellinger,
M.T.,
Lorens,
J.B. &
Brekken,
R.A.
(2015) Warfarin blocks Gas6-mediated axl activation required for pancreatic cancer epithelial plasticity and metastasis. Cancer Res., 75, 3699-3705.
-
Krautler,
V.,
Van Gunsteren,
W.F. &
Hunenberger,
P.H.
(2001) A fast SHAKE: algorithm to solve distance constraint equations for small molecules in molecular dynamics simulations. J. Comput. Chem., 22, 501-508.
-
Lee,
S.Y.,
Lim,
T.G.,
Chen,
H.,
Jung,
S.K.,
Lee,
H.J.,
Lee,
M.H.,
Kim,
D.J.,
Shin,
A.,
Lee,
K.W.,
Bode,
A.M.,
Surh,
Y.J. &
Dong,
Z.
(2013) Esculetin suppresses proliferation of human colon cancer cells by directly targeting beta-catenin. Cancer Prev. Res. (Phila.), 6, 1356-1364.
-
Li,
H.,
Liu,
A.,
Zhao,
Z.,
Xu,
Y.,
Lin,
J.,
Jou,
D. &
Li,
C.
(2011) Fragment-based drug design and drug repositioning using multiple ligand simultaneous docking (MLSD): identifying celecoxib and template compounds as novel inhibitors of signal transducer and activator of transcription 3 (STAT3). J. Med. Chem., 54, 5592-5596.
-
Liu,
G.,
Liu,
Z.,
Yan,
Y. &
Wang,
H.
(2017) Effect of fraxetin on proliferation and apoptosis in breast cancer cells. Oncol. Lett., 14, 7374-7378.
-
Liu,
H.,
An,
X.,
Li,
S.,
Wang,
Y.,
Li,
J. &
Liu,
H.
(2015) Interaction mechanism exploration of R-bicalutamide/S-1 with WT/W741L AR using molecular dynamics simulations. Mol. Biosyst., 11, 3347-3354.
-
Loncharich,
R.J.,
Brooks,
B.R. &
Pastor,
R.W.
(1992) Langevin dynamics of peptides: the frictional dependence of isomerization rates of N-acetylalanyl-N'-methylamide. Biopolymers, 32, 523-535.
-
Maier,
J.A.,
Martinez,
C.,
Kasavajhala,
K.,
Wickstrom,
L.,
Hauser,
K.E. &
Simmerling,
C.
(2015) ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput., 11, 3696-3713.
-
Miller,
B.R. 3rd, McGee, T.D. Jr.,
Swails,
J.M.,
Homeyer,
N.,
Gohlke,
H. &
Roitberg,
A.E.
(2012) MMPBSA.py: an efficient program for end-state free energy calculations. J. Chem. Theory Comput., 8, 3314-3321.
-
Molina-Jimenez,
M.F.,
Sanchez-Reus,
M.I.,
Andres,
D.,
Cascales,
M. &
Benedi,
J.
(2004) Neuroprotective effect of fraxetin and myricetin against rotenone-induced apoptosis in neuroblastoma cells. Brain Res., 1009, 9-16.
-
Morris,
G.M.,
Huey,
R.,
Lindstrom,
W.,
Sanner,
M.F.,
Belew,
R.K.,
Goodsell,
D.S. &
Olson,
A.J.
(2009) AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem., 30, 2785-2791.
-
Peng,
X.M.,
Damu,
G.L. &
Zhou,
C.
(2013) Current developments of coumarin compounds in medicinal chemistry. Curr. Pharm. Des., 19, 3884-3930.
-
Pinto,
D. &
Silva,
A.M.S.
(2017) Anticancer natural coumarins as lead compounds for the discovery of new drugs. Curr. Top. Med. Chem., 17, 3190-3198.
-
Reck,
M.,
Heigener,
D.F.,
Mok,
T.,
Soria,
J.C. &
Rabe,
K.F.
(2013) Management of non-small-cell lung cancer: recent developments. Lancet, 382, 709-719.
-
Roe,
D.R. &
Cheatham,
T.E. 3rd
(2013) PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput., 9, 3084-3095.
-
Rossi,
A. &
Di Maio,
M.
(2016) Platinum-based chemotherapy in advanced non-small-cell lung cancer: optimal number of treatment cycles. Expert Rev. Anticancer Ther., 16, 653-660.
-
Shin,
J.,
Lee,
H.J.,
Jung,
D.B.,
Jung,
J.H.,
Lee,
H.J.,
Lee,
E.O.,
Lee,
S.G.,
Shim,
B.S.,
Choi,
S.H.,
Ko,
S.G.,
Ahn,
K.S.,
Jeong,
S.J. &
Kim,
S.H.
(2011) Suppression of STAT3 and HIF-1 alpha mediates anti-angiogenic activity of betulinic acid in hypoxic PC-3 prostate cancer cells. PLoS One, 6, e21492.
-
Wang,
G.,
Lu,
M.,
Yao,
Y.,
Wang,
J. &
Li,
J.
(2017) Esculetin exerts antitumor effect on human gastric cancer cells through IGF-1/PI3K/Akt signaling pathway. Eur. J. Pharmacol., 814, 207-215.
-
Wang,
H.,
Zou,
D.,
Xie,
K. &
Xie,
M.
(2014) Antibacterial mechanism of fraxetin against Staphylococcus aureus. Mol. Med. Rep., 10, 2341-2345.
-
Wang,
J.,
Wolf,
R.M.,
Caldwell,
J.W.,
Kollman,
P.A. &
Case,
D.A.
(2004) Development and testing of a general amber force field. J. Comput. Chem., 25, 1157-1174.
-
Xiao,
H.,
Bid,
H.K.,
Jou,
D.,
Wu,
X.,
Yu,
W.,
Li,
C.,
Houghton,
P.J. &
Lin,
J.
(2015) A novel small molecular STAT3 inhibitor, LY5, inhibits cell viability, cell migration, and angiogenesis in medulloblastoma cells. J. Biol. Chem., 290, 3418-3429.
-
Xu,
X.,
Liu,
X. &
Zhang,
Y.
(2018) Osthole inhibits gastric cancer cell proliferation through regulation of PI3K/AKT. PLoS One, 13, e0193449.
-
Xu,
X. &
Zhu,
Y.
(2017) Curcumin inhibits human non-small cell lung cancer xenografts by targeting STAT3 pathway. Am. J. Transl. Res., 9, 3633-3641.
-
Xue,
W.,
Yang,
F.,
Wang,
P.,
Zheng,
G.,
Chen,
Y.,
Yao,
X. &
Zhu,
F.
(2018) What contributes to serotonin-norepinephrine reuptake inhibitors’ dual-targeting mechanism? The key role of transmembrane domain 6 in human serotonin and norepinephrine transporters revealed by molecular dynamics simulation. ACS Chem. Neurosci., 9, 1128-1140.