Abstract
Nephronophthisis (NPHP) is an autosomal recessive cystic kidney disease that is characterized by primary ciliary dysfunction (ciliopathy) and progresses to end-stage kidney disease (ESKD) during the second decade of life (juvenile and adolescent NPHP) or before the age of 3 years (infantile NPHP). Here we describe the case of an infant with NPHP who carries a homozygous mutation in SDCCAG8 (also called NPHP10 or BBS16) that encodes SDCCAG8 (serologically defined colon cancer antigen 8). SDCCAG8 is localized at the centrioles of both renal epithelial cells and retinal photoreceptor cells. A mutation in SDCCAG8 is also associated with Bardet-Biedl syndrome (BBS), characterized by NPHP, obesity, polydactyly, and rod-cone dystrophy. A 2-year-old boy was referred to our hospital due to kidney dysfunction of unknown etiology; the patient presented with delayed development and opsoclonus but did not exhibit the clinical characteristics of BBS. Histological findings such as dilatation of tubules and irregular thickness of tubular basement membrane confirmed the diagnosis of NPHP. Four months after referral, the patient’s renal function was rapidly deteriorated, and emergency peritoneal dialysis was initiated. Next-generation sequencing (NGS) was performed, showing that the patient carries a homozygous four-base-pair deletion in SDCCAG8 (c.849_852delTTTG, p.Cys283Ter). The patient’s parents were also found to be heterozygous for this loss-of-function mutation. To the best of our knowledge, the present patient is the first case of biopsy-proven infantile NPHP with a homozygous SDCCAG8 mutation. We conclude that NGS is extremely useful in the identification of SDCCAG8-related NPHP as a cause of sudden-onset ESKD during infancy.
Introduction
Nephronophthisis (NPHP) is an autosomal recessive cystic kidney disease (characterized by tubular basement membrane disintegration, tubular cyst formation, and tubulointerstitial inflammation and fibrosis) that causes end-stage kidney disease (ESKD) in children (Halbritter et al. 2013). With respect to the age of ESKD onset, three clinical variants of NPHP have been described: infantile, juvenile, and adolescent types (Saunier et al. 2005). Infantile NPHP is a rare clinical condition characterized by early-onset ESKD (< 3 years) and is typically associated with mutations in NPHP2 (Gagnadoux et al. 1989). Since the discovery of the first NPHP1 in 1997 (Hildebrandt et al. 1997), 20 different NPHP genes have been identified, and proteins encoded by NPHP genes are localized to the non-motile cilia; thus, NPHP is considered as ciliopathy. Although NPHP generally occurs as an isolated renal disease, approximately 15% of the patients present with extrarenal symptoms such as retinitis pigmentosa or developmental delay (Wolf 2015). If extrarenal symptoms are detected in addition to NPHP, these disorders are categorized as NPHP-related ciliopathy (NPHP-RC).
Bardet-Biedl syndrome (BBS) is a rare type of NPHP-RC characterized by rod–cone dystrophy, polydactyly, obesity, genital malformation, learning difficulties, and renal abnormalities including NPHP (Beales et al. 1999). Recently, mutations in SDCCAG8 (also called BBS16 or NPHP10) that encodes SDCCAG8 (serologically defined colon cancer antigen 8) have been detected in 1%-2% of patients with BBS (Schaefer et al. 2011). SDCCAG8 extends over 2,581 kb on chromosome 1q43-q44 and contains 18 exons. The full-length cDNA of SDCCAG8 encodes an 82.7-kDa protein (713 amino acids) (Otto et al. 2010). SDCCAG8 has been shown to directly interact with oral-facial-digital syndrome 1, a protein that is also associated with NPHP-RC (Otto et al. 2010). Here we describe the first case of biopsy-proven NPHP with a homozygous SDCCAG8 mutation in an infant who experienced rapid ESKD development.
Case Report
A 2-year-old Japanese boy was admitted to our hospital due to kidney dysfunction of unknown etiology. The patient (the first child of healthy nonconsanguineous parents) was born via normal vaginal delivery at 39 weeks and 5 days of gestation with a birth weight of 3,316 g. No family history of renal disease, including NPHP and ESKD, was noted. He had no history of prenatal abnormalities but was diagnosed with delayed development and opsoclonus. At the age of 1 year, he was admitted to a local hospital due to acute pneumonia, leading to the diagnosis of bronchial asthma. At that time, his blood urea nitrogen and serum creatinine levels were 7.2 and 0.3 mg/dL, respectively. Moreover, he had a history of recurrent acute otitis media.
Upon admission, his height was 84.0 cm [−0.5 standard deviation (SD)], body weight was 11.6 kg (+0.0 SD), and blood pressure was 108/62 mmHg. Physical examination findings were normal, except for the presence of opsoclonus. An ophthalmologist examined his eyes and found no structural abnormalities or retinal dystrophy. Moreover, physical abnormalities such as postaxial polydactyly were not observed. The patient had severe developmental delay; thus, he was not able to speak significant words or walk independently. Hematological findings included a white blood cell count of 5,700/μL, with 25.3% neutrophils and 67.4% lymphocytes; a hemoglobin level of 9.4 g/dL; and a platelet count of 17.0 × 104/μL. Meanwhile, the blood biochemistry and serologic findings showed the levels of various parameters as follows: total protein, 6.5 g/dL; albumin, 4.0 g/dL; urea nitrogen, 18 mg/dL; creatinine, 1.01 mg/dL; cystatin C, 2.37 mg/L; sodium, 140 mEq/L; potassium, 3.6 mEq/L; chloride, 113 mEq/L; calcium, 8.7 mg/dL; and inorganic phosphorus, 5.4 mg/dL. Based on the use of serum creatinine and serum cystatin C, the patient’s estimated glomerular filtration rates (eGFRs) were 29.2 mL/min and 36.1 mL/min, respectively. Moreover, urinalysis results revealed a pH of 5.5, a specific gravity of 1.004, and a total protein creatinine level of 0.24 g/g × Cre; occult blood reaction and leukocyte esterase test results were negative. The urinary beta-2-microglobulin level was high at 11,186 (normal: < 250) μg/L. Renal ultrasonography findings showed that the size of the right kidney was 5.94 cm and that of the left kidney was 6.34 cm. Both kidneys were highly echogenic. A 99mTc dimercaptosuccinic acid scintigraphy showed a decrease in bilateral kidney uptake; however, no scars were observed. Voiding cystourethrography findings revealed the absence of vesicoureteral reflux (VUR) or lower urinary tract abnormalities. Brain magnetic resonance imaging findings were unremarkable and did not show the presence of a molar tooth sign.
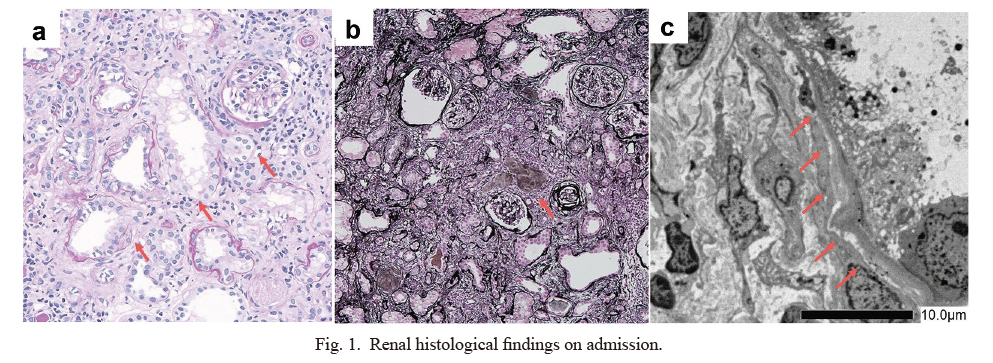
To make a histological diagnosis of kidney dysfunction of unknown etiology, we performed a percutaneous renal biopsy. On light microscopy, no apparent abnormalities were observed in all 33 glomeruli. The interstitium was moderately infiltrated by mononuclear inflammatory cells. The tubules were dilated and contained acellular materials (Fig. 1a, b), Electron microscopy showed dilated tubules and thickening, thinning, and splitting of the tubular basement membrane (Fig. 1c). Based on these histological findings, the patient was diagnosed with infantile NPHP. Despite providing supportive treatment, such as the administration of antihypertensive agents and dietary restrictions, patient’s serum creatinine levels rapidly increased to 4.2 mg/dL. His eGFR was set at 9 mL/min, and he developed anuria. Peritoneal dialysis (PD) was therefore initiated 4 months after renal biopsy. After a 9-month follow-up, the patient’s condition was stable with continuous cycling PD; no catheter-related complications were observed.
A comprehensive genetic analysis of congenital abnormalities in the kidney and urinary tract was performed using next-generation sequencing (NGS). We thus found that the patient carries a homozygous four-base-pair deletion in SDCCAG8 (NM_006642.5; c.849_852delTTTG, p. Cys283Ter), leading to the loss of function that could cause retinal–renal ciliopathy. This four-base-pair deletion was also identified in a single allele of each parent (Fig. 2). Incidentally, the same deletion mutation was already reported in a Japanese girl with BBS (Yamamura et al. 2017).
The ethics committee of Kobe University Graduate School of Medicine approved the genomic analysis (approval number: 301), and a written informed consent was obtained from the patient’s parents. All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional research committee and/or national research committee at which the study was conducted, and the study was conducted in accordance with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
Discussion
The prevalence rate of renal abnormalities, such as renal cysts and VUR, in patients with BBS is approximately 53%-82% (Imhoff et al. 2011; Forsythe et al. 2018). However, only 5%-25% of these patients develop kidney dysfunction and 4%-10% present with progression to ESKD after the second decade of life (Rathi et al. 2007; Forsythe et al. 2018). Thus, although ESKD is the most critical cause of death in BBS, it is not a common characteristic in individuals with BBS. In the last two decades, 21 causal genes (BBS1-21) have been identified in patients with BBS (Forsythe et al. 2018). In terms of causal genes, recent studies have shown that mutations in SDCCAG8 could play an important role in patients with BBS who present with early-onset ESKD but without polydactyly (Schaefer et al. 2011; Billingsley et al. 2012). Our patient presented with the earliest-onset ESKD among all cases reported in the literature (Otto et al. 2010; Schaefer et al. 2011; Billingsley et al. 2012; Yamamura et al. 2017). In addition, although a combination of ESKD and developmental delay was observed in our patient, no other characteristics of BBS, such as early-onset obesity, polydactyly, and rod-cone dystrophy, were identified at the onset of ESKD.
Schaefer et al. (2011) revealed that all patients developed early-onset ESKD (range: 5-28 years) in an analysis of five families with mutations in SDCCAG8 in France and the United States. However, none of the patients had polydactyly, which is present in 58%-74% of all patients with BBS. Moreover, they found that all patients in the French cohort presented with bronchial asthma and a history of recurrent pulmonary infections in early childhood, and most patients (83%) had early-onset obesity. Furthermore, Billingsley et al. (2012) reported five patients with SDCCAG8-related BBS who had retinal and renal involvement but no polydactyly; gradually, obesity and bronchial asthma developed in three and two patients, respectively. Although our patient had bronchial asthma and a history of recurrent otitis media, obesity and rod-cone dystrophy were not observed during the follow-up period, which led us to perform renal biopsy to determine the cause of kidney dysfunction.
Recently, Yamamura et al. (2017) have reported a Japanese girl with BBS who presented with the same homozygous four-base-pair deletion in SDCCAG8 as our patient. However, a histological diagnosis could not be established because kidney biopsy was not performed in their patient. At the age of 3 years and 9 months, emergency PD was initiated due to rapid deterioration in kidney function within 3 months of admission. Although she did not present with obesity and polydactyly, retinitis pigmentosa was identified via ocular examination (Yamamura et al. 2017). Thus, regular examination by an ophthalmologist is necessary for our patient because retinitis pigmentosa may develop later in his life.
To the best of our knowledge, this is the first case of SDCCAG8-related infantile NPHP, diagnosed on the basis of histological findings of dilatation of tubules and irregular thickness of tubular basement membrane. In conclusion, SDCCAG8-related NPHP should be considered as a cause of sudden-onset ESKD in early childhood, even in the absence of obesity and rod–cone dystrophy. NGS is extremely useful in the identification of SDCCAG8 mutations and early diagnosis, thereby preventing the need for kidney biopsy.
We presented a preliminary report of this case at the 122nd Annual Meeting of the Japanese Pediatric Society 2019.
Acknowledgments
S.F. has received clinical research funding B at the Saitama Children’s Medical Center.
Conflict of Interest
The authors declare no conflict of interest.
References
-
Beales,
P.L.,
Elcioglu,
N.,
Woolf,
A.S.,
Parker,
D. &
Flinter,
F.A.
(1999) New criteria for improved diagnosis of Bardet-Biedl syndrome: results of a population survey. J. Med. Genet., 36, 437-446.
-
Billingsley,
G.,
Vincent,
A.,
Deveault,
C. &
Heon,
E.
(2012) Mutational analysis of SDCCAG8 in Bardet-Biedl syndrome patients with renal involvement and absent polydactyly. Ophthalmic Genet., 33, 150-154.
-
Forsythe,
E.,
Kenny,
J.,
Bacchelli,
C. &
Beales,
P.L.
(2018) Managing Bardet-Biedl syndrome-now and in the future. Front. Pediatr., 6, 23.
-
Gagnadoux,
M.F.,
Bacri,
J.L.,
Broyer,
M. &
Habib,
R.
(1989) Infantile chronic tubulo-interstitial nephritis with cortical microcysts: variant of nephronophthisis or new disease entity? Pediatr. Nephrol., 3, 50-55.
-
Halbritter,
J.,
Porath,
J.D.,
Diaz,
K.A.,
Braun,
D.A.,
Kohl,
S.,
Chaki,
M.,
Allen,
S.J.,
Soliman,
N.A.,
Hildebrandt,
F. &
Otto,
E.A.
;
GPN Study Group
(2013) Identification of 99 novel mutations in a worldwide cohort of 1,056 patients with a nephronophthisis-related ciliopathy. Hum. Genet., 132, 865-884.
-
Hildebrandt,
F.,
Strahm,
B.,
Nothwang,
H.G.,
Gretz,
N.,
Schnieders,
B.,
Singh-Sawhney,
I.,
Kutt,
R.,
Vollmer,
M. &
Brandis,
M.
(1997) Molecular genetic identification of families with juvenile nephronophthisis type 1: rate of progression to renal failure. APN Study Group. Arbeitsgemeinschaft fur Padiatrische Nephrologie. Kidney Int., 51, 261-269.
-
Imhoff,
O.,
Marion,
V.,
Stoetzel,
C.,
Durand,
M.,
Holder,
M.,
Sigaudy,
S.,
Sarda,
P.,
Hamel,
C.P.,
Brandt,
C.,
Dollfus,
H. &
Moulin,
B.
(2011) Bardet-Biedl syndrome: a study of the renal and cardiovascular phenotypes in a French cohort. Clin. J. Am. Soc. Nephrol., 6, 22-29.
-
Otto,
E.A.,
Hurd,
T.W.,
Airik,
R.,
Chaki,
M.,
Zhou,
W.,
Stoetzel,
C.,
Patil,
S.B.,
Levy,
S.,
Ghosh,
A.K.,
Murga-Zamalloa,
C.A.,
van Reeuwijk,
J.,
Letteboer,
S.J.,
Sang,
L.,
Giles,
R.H.,
Liu,
Q.,
et al. (2010) Candidate exome capture identifies mutation of SDCCAG8 as the cause of a retinal-renal ciliopathy. Nat. Genet., 42, 840-850.
-
Saunier,
S.,
Salomon,
R. &
Antignac,
C.
(2005) Nephronophthisis. Curr. Opin. Genet. Dev., 15, 324-331.
-
Schaefer,
E.,
Zaloszyc,
A.,
Lauer,
J.,
Durand,
M.,
Stutzmann,
F.,
Perdomo-Trujillo,
Y.,
Redin,
C.,
Bennouna Greene,
V.,
Toutain,
A.,
Perrin,
L.,
Gerard,
M.,
Caillard,
S.,
Bei,
X.,
Lewis,
R.A.,
Christmann,
D.,
et al. (2011) Mutations in SDCCAG8/NPHP10 cause Bardet-Biedl syndrome and are associated with penetrant renal disease and absent polydactyly. Mol. Syndromol., 1, 273-281.
-
Rathi,
M.,
Ganguli,
A.,
Singh,
S.K.,
Kohli,
H.S.,
Gupta,
K.L.,
Sakhuja,
V. &
Jha,
V.
(2007) Bardet-Biedl syndrome with end-stage kidney disease: a case report and review of literature. Indian J. Nephrol., 17, 10-13.
-
Wolf,
M.T.
(2015) Nephronophthisis and related syndromes. Curr. Opin. Pediatr., 27, 201-211.
-
Yamamura,
T.,
Morisada,
N.,
Nozu,
K.,
Minamikawa,
S.,
Ishimori,
S.,
Toyoshima,
D.,
Ninchoji,
T.,
Yasui,
M.,
Taniguchi-Ikeda,
M.,
Morioka,
I.,
Nakanishi,
K.,
Nishio,
H. &
Iijima,
K.
(2017) Rare renal ciliopathies in non-consanguineous families that were identified by targeted resequencing. Clin. Exp. Nephrol., 21, 136-142.