Abstract
Synpolydactyly is a congenital limb malformation characterized by incomplete separation and duplication in fingers and/or toes, which is mainly caused by mutations in the homeobox D13 (HOXD13) gene. Here, a four-generation family with variant phenotypes of synpolydactyly was analyzed, in which the proband had bilateral preaxial synpolydactyly in toes with normal fingers, the father had clinodactyly in the fifth fingers, while the mother and grandma was normal. Trio whole-exome sequencing (trio-WES) is a high throughput sequencing targeting whole genome for detecting exonic variants from the proband and the parents in a family. Through trio-WES followed by Sanger sequencing and enzyme digestion, a heterozygous nonsense mutation (c.859 C>T/p.Gln287Ter) was newly identified in the homeodomain of the HOXD13 gene from the proband and the affected father, but not from the unaffected mother, the unaffected grandma, or the normal control. Mutation Taster, Human Splicing Finder and EX-SKIP predicted that the heterozygous mutation (c.859 C>T) would result in haploinsufficiency of HOXD13 protein through nonsense-mediated mRNA decay (NMD) and splicing abnormality, which might disrupt the integrity and reduce the expression level of the HOXD13 protein (loss-of-function). In short, a heterozygous nonsense mutation in the HOXD13 gene was newly identified in two patients with mild phenotypes of synpolydactyly, which extends the mutation spectrum in HOXD13 gene. Moreover, the findings we presented here deepen our understanding of the clinical consequences of non-syndromic synpolydactyly and may provide a new clue for further studies of the pathogenic mechanism of the mutation that causes aberrant splicing of HOXD13 gene.
Introduction
Synpolydactyly (SPD) is a commonly hereditary digital malformation with webbing of adjacent fingers and toes. It was reported that the incidence of SPD is ~ 3-10 in 10,000 births (Castilla et al. 1980). As a highly heterogeneous developmental deformity, the phenotypes of SPD are very variable between inter- and intra-familial members. Typical SPD involves fusion of 3/4 fingers and 2/3 toes (cutaneous or bony) with partial or complete digital reduplication in the webbing, minor variants and unusual phenotypes are also found in some patients (Brison et al. 2014). SPD may segregate into two forms: syndromic and non-syndromic. Besides deformities in digits, abnormalities in multiple systems and organs are usually associated with syndromic SPD, while non-syndromic SPD only involves the fingers and/or toes. According to affected regions and numbers of the digits, SPD was classified into nine well-characterized types based on the Temtamy-McKusick classification, and most of SPD are non-syndromic and mendelian dominant hereditary diseases (Malik 2012). In general, clinical symptoms in autosomal dominant fashion are relatively mild with variable manifestations and incomplete penetrance.
The molecular mechanism of SPD is still ambiguous and difficult to be explored. Typical SPD is generally caused by aberrant expansions of a 15-residue polyalanine tract in homeobox D13 (HOXD13), while the patients with minor variants and unusual phenotypes are provoked by minor mutations (small deletions/insertions, nonsense and missense mutations) in the homeodomain of HOXD13 (Kurban et al. 2011; Xin et al. 2012; Deng et al. 2017). The other genes that are associated with SPD have also been reported (Johnston and Kirby 1955; Cenani and Lenz 1967; Debeer et al. 2002; Li et al. 2010; Wieczorek et al. 2010; Dimitrov et al. 2010), which makes the relationship between the phenotypes and genotypes of SPD much intricate.
The HOX genes are members of a transcription-factor family with a highly conserved trait, they play an important role in early development (Capecchi 1997). The 39 HOX genes of the family consist of four independent clusters in humans: HOXA, HOXB, HOXC, and HOXD (Scott 1993). Among them, HOXD13 is firstly verified to be associated with developmental disorders of human (Muragaki et al. 1996; Goodman 2002). Limb deformities-related mutations in HOXD13 gene of both humans and mice indicate that HOXD13 plays a critical role during limb development. The HOXD13 gene locates on 2q31.1 of chromosome 2, and comprises two exons which respectively codes two important functional domains: a 15-residue polyalanine tract in exon 1 and homeodomain in exon 2 (DNA binding motif).
Trio whole-exome sequencing (trio-WES) is a high throughput sequencing targeting all coding exons in the genome, trio means a three-person group which contains the proband and the parents from one family. In our study, the proband and the parents from a four-generation family with non-syndromic SPD were recruited for screening exonic variants by trio-WES. Finally, a novel nonsense mutation was identified in the HOXD13 gene of the proband and the father, Furthermore, we sought to predict the molecular mechanism of the pathogenic nature of the novel mutation by bioinformatics analysis of Mutation Taster, Human Splicing Finder (HSF) and EX-SKIP.
Materials and Methods
Participants and samples
The family with non-syndromic SPD was from Fujian province in China, the proband was an inpatient from Fuzhou Second Hospital Affiliated to Xiamen University, the proband and his father were diagnosed based on clinical manifestations, imaging and family history. After the informed consent was obtained from the family members, peripheral blood samples were collected from the members and one normal control. The normal control was from the medical examination center of the hospital. This study was approved by the Ethics Committee of Fuzhou Second Hospital Affiliated to Xiamen University.
Trio whole exome sequencing
Following the procedures of SE Blood DNA Kit (Omega Bio-Tek, CA, USA), DNA was extracted from peripheral blood lymphocytes of the family members and the normal control. Trio-WES was performed on the proband and the parents using genomic DNA (gDNA). Each library of gDNA was mixed with capture probes of xGen®Exome Research Panel v1.0 (IDT Technology, Coralville, IA, USA) for liquid hybridization, according to the manufacturer’s instructions, then targeted regions were enriched and total exon library was constructed. Targeted sequences included all coding exons of ~20 thousand human genes and adjacent intronic sequences (10 bp). Prepared samples were run on a NovaSeq 6000 instrument (PE150, Illumina) with sequence coverage of ≥ 99% for the targeted sequences. Quality controls followed and clean data was finally obtained after the removal of the connectors and low-quality reads from the raw data.
Bioinformatics prediction and data analysis
Online Mendelian Inheritance in Man (OMIM), Swiss-Prot diseases and variants (Swiss-var), the human gene mutation database (HGMD), and ClinVar (http://www.ncbi.nlm.nih.gov/clinvar/) were used to annotate disease-related genes, pathogenic sites, and the minor allele frequency (MAF) of mutations that had been reported. Exclusion criteria for nonpathogenic variants was as follows: (1) allele frequency ≥ 1%; (2) intronic or synonymous variants without splicing effect; (3) variants in either the 5′ or 3′ untranslated region. The pathogenic mutation was confirmed according to the standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (Richards et al. 2015).
Verification by Sanger sequencing and enzyme digestion, pathogenicity analysis for the variants
Disease-related variant filtered from trio-WES was validated by Sanger sequencing and enzyme digestion. Genomic DNA sequence (GenBank NG_008137.1) was picked out as reference sequence. The primers and conditions of the polymerase chain reaction (PCR) amplification were referred as previous study (Goodman et al. 1997). Sanger sequencing was performed on an ABI 3730 XL genetic analyzer (Applied Biosystems; Thermo Fisher Scientific, Inc., Waltham, MA, USA). The fragments of PCR-sequencing included both the polyalanine tract in exon 1 and the mutated fragment in exon 2 of HOXD13 gene. Restriction endonuclease was found through NEB cutter V2.0. Enzyme digestion was based on the instructions of the restriction endonuclease. The products of enzyme digestion were separated on a 2% agarose gel and viewed on a gel imaging system (WD-9413B) (Liuyi Biotechnology, Beijing, China). Mutation Taster (http://www.mutationtaster.org/), HSF (Version 3.1) (http://www.umd.be/HSF3/) and EX-SKIP (http://ex-skip.img.cas.cz/) were further selected for probable pathogenic prediction.
Results
Clinical, imaging, and pedigree investigation
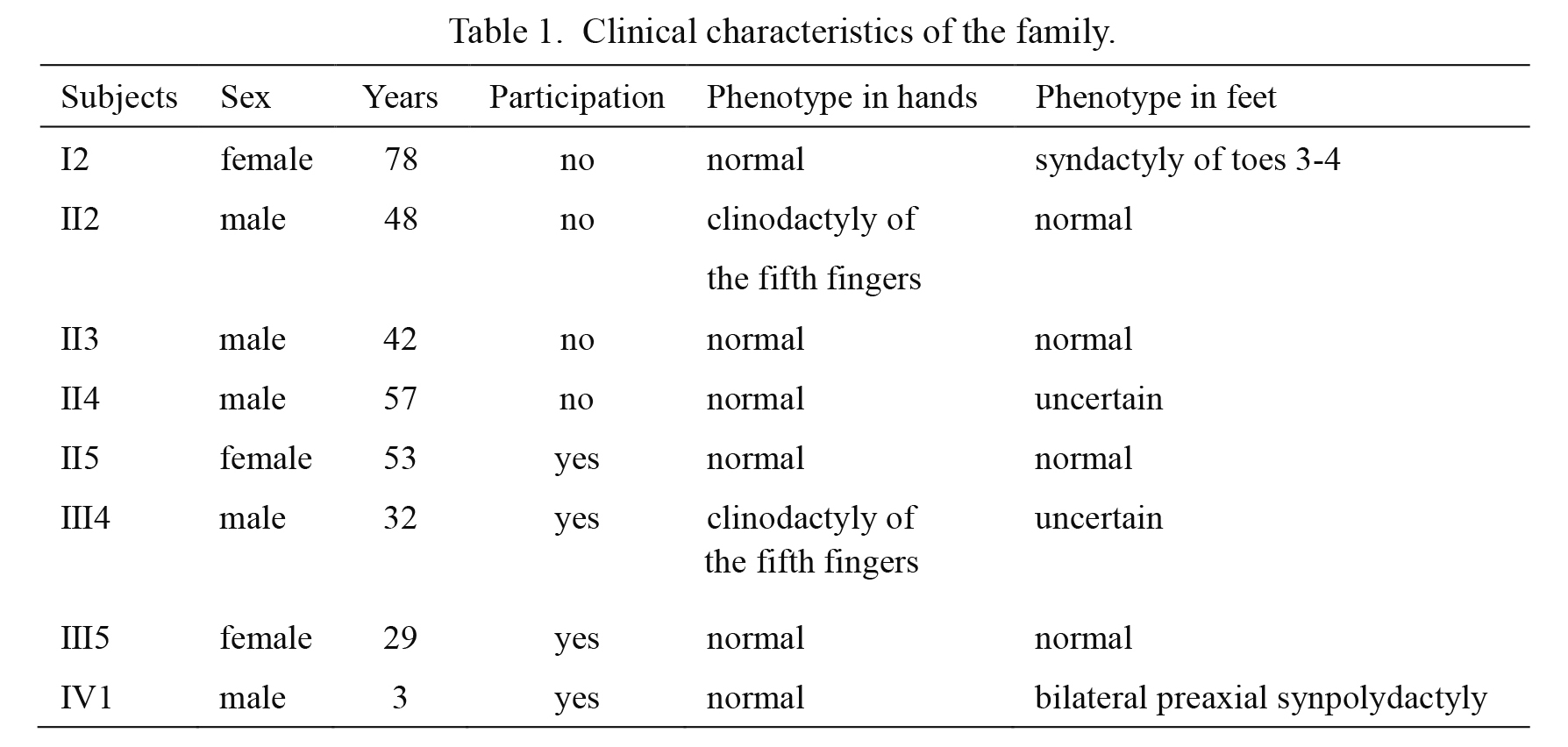
The participants were under detailed physical examination, necessary imaging examination and pedigree investigation by clinicians (Fig. 1). The normal control had simple physical examination. There were four members with obvious and variant phenotypes of SPD, the proband (IV1, Fig. 1) had bilateral preaxial synpolydactyly in toes with normal fingers (Fig. 2A), the father (III4, Fig. 1) had clinodactyly in the fifth fingers (Fig. 2B), the mother and grandma (III5 and II5, Fig. 1) had normal phenotypes, the clinical manifestations of the other members were obtained through pedigree investigation, from which it was found that there were similar manifestations in the second toes between patients III4 and his father (II4, Fig. 1), and thus the clinical presentations of feet in III4 and II4 were ambiguous due to the lacking X-ray diagnosis (Table 1).
Identification of variants by trio-WES and exclusion of irrelevant variants
The affected individuals had different phenotypes of SPD without sex differences, suggesting that the family conformed to the autosomal-dominant model, thus heterozygous variants in both affected patients that were highly related with SPD would be picked out. Finally, four candidate variants from four different genes were picked out according to the phenotypes of the family (Table 2). Considering the hereditary mode and the relationship between the phenotype and genotype of SPD in our study, we assumed that c.859 C>T in HOXD13 gene and c.925C>T in bone morphogenetic protein-4 (BMP4) gene were the most possible mutations that provoked the disease. Nevertheless, c.925C>T in BMP4 gene was responsible for a syndromic disease (Microphthalmia, syndromic 6, OMIM: 607932) and its pathogenic grade from ACMG was uncertain; namely, c.859 C>T in HOXD13 gene was probably the main pathogenic mutation that was responsible for the non-syndromic SPD in our study. The supporting evidence for the pathogenicity of c.859 C>T from ACMG (2015) was as follows: (1) PVS1 (very strong pathogenicity 1): variation may lead to loss of gene function; (2) PM2 (moderate pathogenicity 2): MAF < 0.005; (3) PP3 (supporting pathogenicity 3): Mutation Taster, Genome Evolution Rate Prediction (GERP), phyloP20way, and phastCons20way predicted that the variant affects the gene products and is harmful in the conservativeness and structure of the protein.

Further Sanger sequencing indicated that c.859 C>T was in the exon 2 of HOXD13 gene from both the proband and his father, but not from the mother, the grandma or normal control (Fig. 3A), however, there was no aberrant expansion of polyalanine tract in exon 1 of HOXD13 gene. Because of the nonsense mutation (c.859 C>T) in exon 2 of HOXD13 gene, the protein was truncated at the 287 amino acid (p.Gln287Ter). Meanwhile, through analyzing the fragment by NEB cutter V2.0, it was suggested that a restriction site (HpyCH4V) (5′-TG↓CA-3′) would disappear if c.859 C>T was in the exon 2 of HOXD13 gene (Fig. 4A). After enzymatic digestion of the normal fragment and mutated fragment (335 bp) overnight, two new fragments (133 bp/202 bp) were produced in nearly half of the PCR products from both the proband and the father, instead, the PCR products were almost completely digested into two smaller fragments (133 bp/202 bp) by HpyCH4V in the mother and normal control (Fig. 4B).

Mutation Taster predicted that c.859 C>T in HOXD13 gene was a disease-causing mutation through two possible pathogenic mechanisms: nonsense-mediated mRNA decay (NMD) and splicing abnormality; HSF found that a new exonic splicing silencer (ESS) site (5′-GTAGCT-3′) (the underlined letter was the mutated site) would be produced in exon 2 of HOXD13 gene due to the mutation, which may affect the effective splicing of exon 2; EX-SKIP suggested that because of the mutation, the number of ESS in exon 2 of HOXD13 gene would increase by 3 (48→51), while the number of exonic splicing enhancers (ESEs) would decrease by 4 (297→293), which result in a higher chance of exon 2 skipping in HOXD13 gene of the patients (Table 3). According to the prediction from the three bioinformatics tools, it was indicated that the integrity and expression level of HOXD13 protein might be affected by c.859 C>T.
Discussion
SPD is a genetic deformity that is highly heterogeneous in clinical manifestation; in addition to typical symptoms, some patients display minor variants and unusual phenotypes, such as reduced typical manifestations (isolated synpolydactyly in 4/5 toes or 2/3 toes), brachydactyly, clinodactyly, camptodactyly, and duplicated metatarsals in different digits (Malik and Grzeschik 2008; Brison et al. 2014;). These patients are more often found with deletion/insertion, nonsense and missense mutations in HOXD13 gene (Goodman et al. 1998; Kan et al. 2003; Zhao et al. 2007; Fantini et al. 2009). In this paper, the family members had atypical deformities in the fingers and toes, and a heterozygous nonsense mutation (c.859 C>T/p.Gln287Ter) was detected in HOXD13 gene from the proband and his father, which conformed to the mode of autosomal dominant inheritance (Figs. 2 and 3A).
After clinical examination of the participants, we show that all the patients belong to non-syndromic SPD, and the symptoms of clinodactyly of the fifth fingers in patients III4 and II2 had been reported as minor variants of SPD in previous studies (Goodman et al. 1997; Brison et al. 2014; Dai et al. 2014), bilateral preaxial polydactyly in the proband (IV1) had been classified as unusual symptom (Malik and Grzeschik 2008); yet the syndactyly of toes 3-4 in patient I2 was hardly reported in patients elsewhere especially from China with non-syndromic SPD (Fig. 1, Table 1). However, it was present in some rare syndromic diseases, such as Greig cephalopolysynpolydactyly (GCPS) (OMIM 175700) (Debeer et al. 2007). Nevertheless, there were no craniofacial and hand malformations in the patients of our study, though they had the same hereditary mode with GCPS. Considering the distinct variations in clinical manifestations and the genetic model of the family in our study, we suggested that the phenotype in patient I2 may be an unusual trait and novel phenotype of non-syndromic SPD (Fig. 1). It is worth noting that the father of patient III4 (II4, Fig. 1) had similar manifestations in the second toes with patients III4 (Fig. 2), and thus the man (II4) is probably affected with SPD, according to the autosomal dominant inheritance pattern, and the clinical symptoms of feet in II4 and III4 become uncertain for lacking radiography (Table 1).
WES covers the coding regions and adjacent sequences (within 10 bp) of all exons from ~ 20 thousand of functional genes in human, yet WES is only suitable for screening point mutation and small insertion/deletion mutation (within 10 bp), so large deletion/duplication (more than 10 bp), the poly sequence and tandem repetitive sequence (TRS) cannot be detected by WES. In our study, a heterozygous variant (c.859 C>T/p.Gln287 Ter) was detected by trio-WES from a family with non-syndromic SPD, and the mutation was confirmed to be the most probably pathogenic mutation in the family (Figs. 3 and 4), the main supporting evidences were the following three points: (1) biological pathogenic grade from the guide of ACMG (2015): pathogenic (PVS1+PM2+PP3); (2) c.859 C>T/p.Gln287 in HOXD13 gene was found in both the proband and his affected father, but not in the normal mother, grandma, or normal control, which highly conformed to the pattern of autosomal dominant inheritance; (3) The mutation was found in the gene of HOXD13, which was responsible for limb deformities and played an important role in limb development in both humans and mice (Zhao et al. 2007; Salsi et al. 2008; Kuss et al. 2009). Nevertheless, c.925C>T in BMP4 gene was found in the proband and the father, which also fit the autosomal dominant inheritance pattern and was associated with SPD. BMP4 is the pathogenic gene of Microphthalmia, syndromic 6, which mainly features ocular malformation and digit deformities, such as poly/syndactyly (Table 2); some families or patients with polydactyly or other digit abnormalities as part symptoms had been reported with frameshifting mutation or missense mutation (c.278A>G) in BMP4 gene (Bakrania et al. 2008). It was reported that both interdigital webbing/syndactyly and extra digits/polydactyly were provoked by the downregulation of BMP4 expression in distal mesodermal (Naruse et al. 2007). Moreover, BMP4 expression was up-regulated by HOXD13 overexpression (gain-of-function) in vivo (Salsi et al. 2008), and a missense mutation (c.278A>G) in BMP4 gene was found in a patient without syndactyly or polydactyly (only with broad hands, low placed thumbs, and dysplastic nails) in a previous study (Bakrania et al. 2008); therefore, the missense mutation (c.925C>T) in BMP4 gene of the family in our study was probably not the major factor, whereas the nonsense mutation in the HOXD13 gene (c.859 C>T) would down-regulate the BMP4 expression due to the functional haploinsufficiency (loss-of-function mutation), thus c.859 C>T in HOXD13 gene was probably the major cause that triggered SPD through downregulation of BMP4 expression in our study.
Currently, aberrant polyalanine expansion in the polyalanine tract of the HOXD13 gene was reported in numerous unrelated families with classical phenotypes of SPD (Kjaer et al. 2005; Horsnell et al. 2006; Gong et al. 2011; Xin et al. 2012), and it leads to misfolding, degradation or cytoplasmic aggregation of those mutant products through dominant negative effect (Brison et al. 2014). Loss-of-function mutations in the homeodomain (in exon 2 of HOXD13) were usually found in the patients with minor variants and unusual phenotypes of SPD (Kan et al. 2003; Kurban et al. 2011; Dai et al. 2014; Deng et al. 2017). Functional haploinsufficiency caused by truncating mutations in HOXD13 is almost the most plausible mechanism. The integrity of the homeodomain of HOXD13 is damaged due to the mutation, and then the level of the protein with integrated ability to bind DNA is partially reduced through NMD, which would result in haploinsufficiency. In order to overcome the shortcomings of WES that cannot detect the aberrant polyalanine expansion in HOXD13 gene of the family, PCR-sequencing of the polyalanine tract in exon 1 had been also executed afterwards to exclude the most common reason. Finally, a hitherto unknown nonsense mutation (c.859 C>T/p.Gln287Ter) was found in the homeodomain of HOXD13 protein (Fig. 3B); it introduces a stop code at the 287 amino acid and truncates the protein. Thus, the function of binding DNA would be destroyed. Furthermore, several online bioinformatics tools predicted that except for NMD, the nonsense mutation would provoke the disease by aberrant splicing through increasing the number of ESS and providing a higher chance of exon 2 skipping. Thus, the expressional level of HOXD13 protein with integrated domains would be reduced and the capacity of DNA binding would decrease, which provided a new clue for the molecular mechanism of SPD. Meanwhile, it was coincident with the conclusion that exonic mutations would affect pre-mRNA splicing in some diseases (Cartegni et al. 2002; Santoro et al. 2007).
In conclusion, a heterozygous nonsense mutation in the HOXD13 gene was identified by trio-WES in a four-generation family with non-syndromic synpolydactyly. Meanwhile, our pathogenicity prediction may provide a new clue for further studies of the pathogenic mechanism of the mutation in the exonic splicing regulatory of the HOXD13 gene.
Acknowledgments
This work was supported by Youth Research Project of Health and Family Planning in Fujian Province (2016-2-31 and 2016-2-41), and Project Funded by Natural Science Foundation of Fujian Province (2019J01541).
Conflict of Interest
The authors declare no conflict of interest
References
-
Bakrania,
P.,
Efthymiou,
M.,
Klein,
J.C.,
Salt,
A.,
Bunyan,
D.J.,
Wyatt,
A.,
Ponting,
C.P.,
Martin,
A.,
Williams,
S.,
Lindley,
V.,
Gilmore,
J.,
Restori,
M.,
Robson,
A.G.,
Neveu,
M.M.,
Holder,
G.E.,
et al. (2008) Mutations in BMP4 cause eye, brain, and digit developmental anomalies: overlap between the BMP4 and hedgehog signaling pathways. Am. J. Hum. Genet., 82, 304-319.
-
Brison,
N.,
Debeer,
P. &
Tylzanowski,
P.
(2014) Joining the fingers: a HOXD13 Story. Dev. Dyn., 243, 37-48.
-
Capecchi,
M.R.
(1997) Hox genes and mammalian development. Cold Spring Harb. Symp. Quant. Biol., 62, 273-281.
-
Cartegni,
L.,
Chew,
S.L. &
Krainer,
A.R.
(2002) Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nat. Rev. Genet., 3, 285-298.
-
Castilla,
E.E.,
Paz,
J.E. &
Orioli-Parreiras,
I.M.
(1980) Syndactyly: frequency of specific types. Am. J. Med. Genet., 5, 357-364.
-
Cenani,
A. &
Lenz,
W.
(1967) Total syndactylia and total radioulnar synostosis in 2 brothers. A contribution on the genetics of syndactylia. Z. Kinderheilkd., 101, 181-190.
-
Dai,
L.,
Liu,
D.,
Song,
M.,
Xu,
X.,
Xiong,
G.,
Yang,
K.,
Zhang,
K.,
Meng,
H.,
Guo,
H. &
Bai,
Y.
(2014) Mutations in the homeodomain of HOXD13 cause syndactyly type 1-c in two Chinese families. PLoS One, 9, e96192.
-
Debeer,
P.,
Devriendt,
K.,
De Smet,
L.,
Deravel,
T.,
Gonzalez-Meneses,
A.,
Grzeschik,
K.H. &
Fryns,
J.P.
(2007) The spectrum of hand and foot malformations in patients with Greig cephalopolysyndactyly. J. Child. Orthop., 1, 143-150.
-
Debeer,
P.,
Schoenmakers,
E.F.,
Twal,
W.O.,
Argraves,
W.S.,
De Smet,
L.,
Fryns,
J.P. &
Van De Ven,
W.J.
(2002) The fibulin-1 gene (FBLN1) is disrupted in a t(12;22) associated with a complex type of synpolydactyly. J. Med. Genet., 39, 98-104.
-
Deng,
H.,
Tan,
T.,
He,
Q.,
Lin,
Q.,
Yang,
Z.,
Zhu,
A.,
Guan,
L.,
Xiao,
J.,
Song,
Z. &
Guo,
Y.
(2017) Identification of a missense HOXD13 mutation in a Chinese family with syndactyly type I-c using exome sequencing. Mol. Med. Rep., 16, 473-477.
-
Dimitrov,
B.I.,
Voet,
T.,
De Smet,
L.,
Vermeesch,
J.R.,
Devriendt,
K.,
Fryns,
J.P. &
Debeer,
P.
(2010) Genomic rearrangements of the GREM1-FMN1 locus cause oligosyndactyly, radio-ulnar synostosis, hearing loss, renal defects syndrome and Cenani-Lenz-like non-syndromic oligosyndactyly. J. Med. Genet., 47, 569-574.
-
Fantini,
S.,
Vaccari,
G.,
Brison,
N.,
Debeer,
P.,
Tylzanowski,
P. &
Zappavigna,
V.
(2009) A G220V substitution within the N-terminal transcription regulating domain of HOXD13 causes a variant synpolydactyly phenotype. Hum. Mol. Genet., 18, 847-860.
-
Gong,
L.,
Wang,
B.,
Wang,
J.,
Yu,
H.,
Ma,
X. &
Yang,
J.
(2011) Polyalanine repeat expansion mutation of the HOXD13 gene in a Chinese family with unusual clinical manifestations of synpolydactyly. Eur. J. Med. Genet., 54, 108-111.
-
Goodman,
F.,
Giovannucci-Uzielli,
M.L.,
Hall,
C.,
Reardon,
W.,
Winter,
R. &
Scambler,
P.
(1998) Deletions in HOXD13 segregate with an identical, novel foot malformation in two unrelated families. Am. J. Hum. Genet., 63, 992-1000.
-
Goodman,
F.R.
(2002) Limb malformations and the human HOX genes. Am. J. Med. Genet., 112, 256-265.
-
Goodman,
F.R.,
Mundlos,
S.,
Muragaki,
Y.,
Donnai,
D.,
Giovannucci-Uzielli,
M.L.,
Lapi,
E.,
Majewski,
F.,
McGaughran,
J.,
McKeown,
C.,
Reardon,
W.,
Upton,
J.,
Winter,
R.M.,
Olsen,
B.R. &
Scambler,
P.J.
(1997) Synpolydactyly phenotypes correlate with size of expansions in HOXD13 polyalanine tract. Proc. Natl. Acad. Sci. USA, 94, 7458-7463.
-
Horsnell,
K.,
Ali,
M.,
Malik,
S.,
Wilson,
L.,
Hall,
C.,
Debeer,
P. &
Crow,
Y.
(2006) Clinical phenotype associated with homozygosity for a HOXD13 7-residue polyalanine tract expansion. Eur. J. Med. Genet., 49, 396-401.
-
Johnston,
O. &
Kirby,
V.V. Jr.
(1955) Syndactyly of the ring and little finger. Am. J. Hum. Genet., 7, 80-82.
-
Kan,
S.H.,
Johnson,
D.,
Giele,
H. &
Wilkie,
A.O.
(2003) An acceptor splice site mutation in HOXD13 results in variable hand, but consistent foot malformations. Am. J. Med. Genet. A, 121A, 69-74.
-
Kjaer,
K.W.,
Hansen,
L.,
Eiberg,
H.,
Utkus,
A.,
Skovgaard,
L.T.,
Leicht,
P.,
Opitz,
J.M. &
Tommerup,
N.
(2005) A 72-year-old Danish puzzle resolved: comparative analysis of phenotypes in families with different-sized HOXD13 polyalanine expansions. Am. J. Med. Genet. A, 138, 328-339.
-
Kurban,
M.,
Wajid,
M.,
Petukhova,
L.,
Shimomura,
Y. &
Christiano,
A.M.
(2011) A nonsense mutation in the HOXD13 gene underlies synpolydactyly with incomplete penetrance. J. Hum. Genet., 56, 701-706.
-
Kuss,
P.,
Villavicencio-Lorini,
P.,
Witte,
F.,
Klose,
J.,
Albrecht,
A.N.,
Seemann,
P.,
Hecht,
J. &
Mundlos,
S.
(2009) Mutant Hoxd13 induces extra digits in a mouse model of synpolydactyly directly and by decreasing retinoic acid synthesis. J. Clin. Invest., 119, 146-156.
-
Li,
Y.,
Pawlik,
B.,
Elcioglu,
N.,
Aglan,
M.,
Kayserili,
H.,
Yigit,
G.,
Percin,
F.,
Goodman,
F.,
Nurnberg,
G.,
Cenani,
A.,
Urquhart,
J.,
Chung,
B.D.,
Ismail,
S.,
Amr,
K.,
Aslanger,
A.D.,
et al. (2010) LRP4 mutations alter Wnt/beta-catenin signaling and cause limb and kidney malformations in Cenani-Lenz syndrome. Am. J. Hum. Genet., 86, 696-706.
-
Malik,
S.
(2012) Syndactyly: phenotypes, genetics and current classification. Eur. J. Hum. Genet., 20, 817-824.
-
Malik,
S. &
Grzeschik,
K.H.
(2008) Synpolydactyly: clinical and molecular advances. Clin. Genet., 73, 113-120.
-
Muragaki,
Y.,
Mundlos,
S.,
Upton,
J. &
Olsen,
B.R.
(1996) Altered growth and branching patterns in synpolydactyly caused by mutations in HOXD13. Science, 272, 548-551.
-
Naruse,
T.,
Takahara,
M.,
Takagi,
M.,
Oberg,
K.C. &
Ogino,
T.
(2007) Busulfan-induced central polydactyly, syndactyly and cleft hand or foot: a common mechanism of disruption leads to divergent phenotypes. Dev. Growth Differ., 49, 533-541.
-
Richards,
S.,
Aziz,
N.,
Bale,
S.,
Bick,
D.,
Das,
S.,
Gastier-Foster,
J.,
Grody,
W.W.,
Hegde,
M.,
Lyon,
E.,
Spector,
E.,
Voelkerding,
K. &
Rehm,
H.L.
(2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med., 17, 405-424.
-
Salsi,
V.,
Vigano,
M.A.,
Cocchiarella,
F.,
Mantovani,
R. &
Zappavigna,
V.
(2008) Hoxd13 binds in vivo and regulates the expression of genes acting in key pathways for early limb and skeletal patterning. Dev. Biol., 317, 497-507.
-
Santoro,
M.,
Modoni,
A.,
Sabatelli,
M.,
Madia,
F.,
Piemonte,
F.,
Tozzi,
G.,
Ricci,
E.,
Tonali,
P.A. &
Silvestri,
G.
(2007) Chronic GM2 gangliosidosis type Sandhoff associated with a novel missense HEXB gene mutation causing a double pathogenic effect. Mol. Genet. Metab., 91, 111-114.
-
Scott,
M.P.
(1993) A rational nomenclature for vertebrate homeobox (HOX) genes. Nucleic Acids Res., 21, 1687-1688.
-
Wieczorek,
D.,
Pawlik,
B.,
Li,
Y.,
Akarsu,
N.A.,
Caliebe,
A.,
May,
K.J.,
Schweiger,
B.,
Vargas,
F.R.,
Balci,
S.,
Gillessen-Kaesbach,
G. &
Wollnik,
B.
(2010) A specific mutation in the distant sonic hedgehog (SHH) cis-regulator (ZRS) causes Werner mesomelic syndrome (WMS) while complete ZRS duplications underlie Haas type polysyndactyly and preaxial polydactyly (PPD) with or without triphalangeal thumb. Hum. Mutat., 31, 81-89.
-
Xin,
Q.,
Li,
L.,
Li,
J.,
Qiu,
R.,
Guo,
C.,
Gong,
Y. &
Liu,
Q.
(2012) Eight-alanine duplication in homeobox D13 in a Chinese family with synpolydactyly. Gene, 499, 48-51.
-
Zhao,
X.,
Sun,
M.,
Zhao,
J.,
Leyva,
J.A.,
Zhu,
H.,
Yang,
W.,
Zeng,
X.,
Ao,
Y.,
Liu,
Q.,
Liu,
G.,
Lo,
W.H.,
Jabs,
E.W.,
Amzel,
L.M.,
Shan,
X. &
Zhang,
X.
(2007) Mutations in HOXD13 underlie syndactyly type V and a novel brachydactyly-syndactyly syndrome. Am. J. Hum. Genet., 80, 361-371.