Abstract
Vascular calcification is a typical feature of atherosclerosis and is associated with adverse cardiovascular events such as myocardial infarction and stroke. Several studies have suggested that adenosine, an ATP metabolite may function as an endogenous regulator of arterial calcification. However, its effects on vascular smooth muscle cell calcification have not been clarified. In this study, we investigated the inhibitory effects of adenosine on vascular calcification in vitro by utilizing the culture of human aortic smooth muscle cells (HASMCs). Osteoblastic differentiation of HASMCs was induced by the treatment with oncostatin M and osteogenic differentiation medium. Adenosine and its metabolically stable analogue, 2-chloroadenosine (CADO) significantly reduced matrix mineralization and alkaline phosphatase (ALP) activities in HASMCs. The mRNA expression of tissue non-specific alkaline phosphatase (TNAP) was down-regulated by adenosine and CADO, but the mRNA expression of other osteoblastic differentiation markers, such as Runt-related transcription factor 2 (RUNX2) and bone sialoprotein (BSP)-II, was not significantly affected by these two reagents. Among the adenosine receptor (AR) subtype-selective agonists used, only IB-MECA (A3 AR-selective agonist) significantly decreased in vitro mineralization and ALP activities in HASMCs, but not with CCPA (A1 AR-selective agonist), CGS21680 (A2a AR-selective agonist), or BAY60-6583 (A2b AR-selective agonist). Importantly, IB-MECA also down-regulated expression of TNAP mRNA. Finally, knockdown of A3 AR, but not A1 AR, A2a AR, or A2b AR, significantly reversed the inhibitory actions of adenosine, CADO, or IB-MECA on in vitro calcification and ALP activities in HASMCs. These data suggest that adenosine attenuates HASMC calcification through A3 AR.
Introduction
Vascular calcification is a prominent feature of chronic inflammatory disorders such as type 2 diabetes, chronic kidney disease, and atherosclerosis and is associated with an increased risk for adverse cardiovascular events (Wilson et al. 2001; Santos et al. 2010; Budoff et al. 2011; Chen et al. 2017). Although the precise mechanisms of ectopic mineralization within the arterial wall remain to be clarified, the concept that vascular calcification is an active and regulated process analogous to osteogenesis is now generally accepted (Demer 1995; Shioi and Ikari 2018). Phenotypic changes of vascular smooth muscle cells (VSMCs) into osteoblast-like cells play a key role in the development of vascular calcification (Leopold 2015; Durham et al. 2018; Hortells et al. 2018). This phenomenon can be induced by proinflammatory cytokines such as TNF-α and IL-1β produced by macrophages infiltrated both in intimal and medial layers of the arteries (Tintut et al. 2000; Ceneri et al. 2017; Benz et al. 2018). We have also shown that oncostatin M (OSM), an IL-6 type cytokine derived from activated macrophages accelerates osteogenic differentiation of human VSMC (HVSMC) through JAK3-STAT3 pathway (Shioi et al. 2002; Kakutani et al. 2015) as compared with other HVSMC calcification models utilizing TNF-α (Kawada et al. 2018).
Adenosine is an essential regulator of vascular homeostasis and exerts its effects via binding to four adenosine receptor (AR) subtypes such as A1, A2a, A2b, and A3. A1 and A3 ARs inhibit adenylyl cyclase (AC), whereas A2a and A2b ARs stimulate AC and increase intracellular cAMP levels (Borea et al. 2015). Genetic analysis of familial arterial calcification of the lower limb has suggested that adenosine may be a putative endogenous inhibitor of arterial calcification (St Hilaire et al. 2011). Affected members have nonfunctional mutations in the 5′-ectonucleotidase (NT5E) gene which encodes the CD73 protein, an enzyme converting AMP to adenosine and skin fibroblasts derived from these individuals show increased activity of tissue non-specific alkaline phosphatase (TNAP) and in vitro calcification capacity (St Hilaire et al. 2011; Fish et al. 2013). Therefore, it is likely that adenosine may serve as an endogenous inhibitor of vascular calcification through regulating TNAP expression. However, its inhibitory effects on VSMC calcification have not been clarified. Furthermore, the direct actions of adenosine on osteoblastic differentiation of VSMC induced by inflammatory mediators such as TNF-α and OSM have not been explored. In this study, we investigated the inhibitory effects of adenosine signaling on human aortic vascular smooth muscle cell (HASMC) calcification.
Materials and Methods
Reagents
Adenosine (ab120498), CCPA (ab120434), and CGS21680 (ab120453) were obtained from Abcam (Cambridge, UK). 2-Chloroadenosine (CADO) (#3136) and BAY60-6583 (#4472) were purchased from Tocris Bioscience (Bristol, UK). IB-MECA (#1066) was obtained from R&D Systems (Minneapolis, MN).
Cell culture and in vitro differentiation
HASMCs were obtained from KURABO (Osaka, Japan), and maintained in SMC growth medium (Smooth Muscle Cell Basal Medium) [HuMedia-SB2] supplemented with growth factors [HuMedia-SG] containing 5% FBS, 0.5 ng/ml of hEGF, 2 ng/ml of hFGF-B, 5 μg/ml of insulin, 50 μg/ml of gentamicin, and 50 ng/ml of amphotericin B). The cells were plated at 1.2 × 105 cells/well in 12-well plates. After confluency, in order to induce osteoblastic differentiation, HASMCs were incubated for the indicated period of time with 10 ng/ml oncostatin M (OSM) (R&D Systems) and osteogenic differentiation medium (ODM) containing 10% FBS, 0.1 μM dexamethasone, 50 μg/ml ascorbic acid 2-phosphate, and 10 mM β-glycerophosphate in Dulbecco’s modified Eagle medium (DMEM).
Alkaline phosphatase (ALP) assay
HASMCs were cultured for the indicated period of time, washed twice with PBS, lysed with 1% Triton X-100 in 0.9% NaCl, and centrifuged. ALP activity of the supernatants was assayed as previously described (Shioi et al. 2002) and the protein concentration was measured using a BCA protein assay kit (Thermo Fisher Scientific, Waltham, MA). The activity to produce 1 nmol of p-nitrophenol was defined as one unit and the values of ALP activities were expressed as units/mg protein.
Alizarin Red S staining and quantification of HASMC calcification
HASMCs were cultured for 8 days in the presence of OSM and ODM. The calcium deposition was visualized by Alizarin Red S staining. The cells were fixed with 4% paraformaldehyde for 10 minutes and then stained with 1% Alizarin Red S/NH4OH (pH 6.4) for 5 minutes. Calcium contents deposited in HASMCs were quantified by methylxylenol blue (MXB) method (Calcium E-test Wako; Wako, Osaka, Japan) and normalized by protein concentrations as previously described (Shioi et al. 2002).
RT-PCR and real-time quantitative RT-PCR
Total RNA of HASMCs was extracted using Sepasol-RNA I Super G (Nacalai tesque, Kyoto, Japan) and cDNAs were reverse synthesized from 1 μg of total RNA using High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA). PCR was carried out in 15 μL of reaction mixture containing 50 ng cDNA, 0.4 μM each primer, 1X GoTaq Green MasterMix (Promega, Madison, WI), and 5% DMSO. We used primer sets of the following genes (sense and anti-sense sequence, respectively): A1 AR (5′-TGC ACT GGC CTG TTC TGT AG-3′, 5′-CTG CCT CTC CCA CGT ACA AT-3′); A2a AR (5′-GGA GTT TGC CCC TTC CTA AG-3′, 5′-CTG CTT CCT CAG AAC CCA AG-3′); A2b AR (5′-ATC TCC AGG TAT CTT CTC-3′, 5′-GTT GGC ATA ATC CAC ACA G-3′); A3 AR (5′-CCT TCT CGC GTG TCC TGA CT-3′, 5′-CTC TGA CTA CCG CCG TTG CT-3′). Thermal cycling condition was set as pre-denaturing at 94°C for 3 minutes, denaturing at 94°C for 30 seconds, annealing at 58°C for 1 minute, extension at 72°C for 1 minute, repeat for 35 cycles, and a final extension at 72°C for 2 minutes.
For real-time quantitative RT-PCR, total RNA of HASMCs and cDNAs were prepared as described above. The mRNA expression levels were quantitatively analyzed by real time RT-PCR using an Applied Biosystems 7500 Fast Real-Time PCR system (Applied Biosystems) and TaqMan Gene Expression Assay (Applied Biosystems). The mRNA expression values were normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA. The primers utilized were as follows: ALPL (TNAP) (Hs00758162_m1), IBSP (bone sialoprotein (BSP)-II) (Hs00173720_m1), Runt-related transcription factor 2 (RUNX2) (Hs00231692_m1), ADORA1 (Hs00186586_g1), ADORA2a (Hs00169123_m1), ADORA2b (Hs00386497_m1) and GAPDH (#402869).
Western blot analysis
Cell lysates were electrophoresed on SDS/polyacrylamide gel and transferred to Immobilon-P membrane (Millipore, Danvers, MA). After blocking, membranes were incubated with the following primary antibodies. Anti-adenosine A3 receptor (ab197350, Abcam) was used at dilution with 1:1,000 and anti-alpha-tubulin (11224-1-AP, Proteintech, Chicago, IL) antibody was used with 1:10,000. After incubation with each corresponding secondary antibody, the signals were detected by enhanced ECL Select™ Western Blotting Detection Reagent (GE Healthcare Amersham, Buckingham, UK).
Small interfering RNA transfections
The FlexiTube GeneSolution siRNAs for A1 AR, A2a AR, A2b AR and Negative Control siRNA (#1022076) were obtained from Qiagen (Hilden, Germany). ON-TARGETplus Human ADORA3 (adenosine A3 receptor) siRNA (L-005418-00) and ON-TARGETplus Non-targeting Control Pool (D-001810-10) were obtained from GE Healthcare Dharmacon (Lafayette, CO). Target sequences of each siRNA were as follows: A1 AR, 5′-GAGGCCCATGTGACTAATAAA-3′, 5′-CAGCATGGAGTACATGGTCTA-3′, 5′-CTGGAGTGTAATTACCTGTCA-3′, 5′-AAGGGTAGGTTCAGTAATCAT-3′; A2a AR, 5′-CCACACCAATTCGGTTGTGAA-3′, 5′-AAGGGCTTGGGTTCTGAGGAA-3′, 5′-CAGCATGAGGCCCAGCAAGAA-3′, 5′-CAGGAGTGTCCTGATGATTCA″; A2b AR, 5′-AAGGATTGACAAATATATTTA-3′, 5′-CACGTATCTAGCTAATATGTA-3′, 5′-TCGGTGTGGGCCTATGATCTA-3′, 5′-AACAGCTTGAATGGATTCTAA-3′; A3 AR, 5′-GGUCAUGCCUUUGGCCAUU-3′, 5′-GGUCAAGCUUACCGUCAGA-3′, 5′-UGACUUGCCUACUGCUUAU-3′, 5′-GCAUCACAAUCCACUUCUA-3′. Cells were treated with antibiotic-free medium containing 10 pmol/mL siRNA using Lipofectamine RNAiMAX (Invitrogen) prepared in OptiMEM (Invitrogen). After 24 hours, the cells were incubated in antibiotic-free medium without siRNA for additional 48 hours and utilized for the experiments.
Statistics
Data were presented as the mean ± SEM and analyzed for statistical significance by one-way ANOVA with post hoc analysis (Tukey’s test). These analyses were performed using the statistics add-in software EZR (Saitama Medical Center, Jichi Medical University, Tochigi, Japan) (Kanda 2013). The values of P < 0.05 were considered statistically significant.
Results
Adenosine and CADO attenuate in vitro calcification and TNAP expression in HASMCs
To examine the effects of adenosine on vascular calcification, we utilized an in vitro model of vascular calcification in which HASMCs underwent osteoblastic differentiation and matrix mineralization by the treatment with OSM and ODM (Kakutani et al. 2015). Matrix mineralization was confirmed by alizarin red S staining in the cultures of HASMCs (Fig. 1A). The calcium deposits firstly developed in the cultures around day 6 and thereafter increased in a time-dependent manner (Fig. 1B). The markers of osteoblastic differentiation such as RUNX2, TNAP, and BSP-II were upregulated in HASMCs in a time-dependent manner (Fig. 1C).
We first examined the effects of adenosine per se in this model by the daily addition of the compound, because adenosine is rapidly metabolized. Mineralization of HASMCs was obviously reduced by adenosine (Fig. 2A) and its effects were dose-dependent (Fig. 2B). Adenosine significantly inhibited ALP activities of HASMCs at the concentrations of 10 and 100 μM (Fig. 2C). Adenosine at 100 μM significantly decreased time-dependent inductions of ALP activities of HASMCs until day 8 (Fig. 2D). Moreover, OSM-induced TNAP mRNA expression was significantly suppressed by adenosine at 10 and 100 μM (Fig. 2E), while it did not affect the expression of RUNX2 and BSP-II (Fig. 2E). To further confirm the inhibitory effects of adenosine signaling in this model, we next utilized a metabolically stable analogue, CADO. CADO significantly decreased HASMC calcification and ALP activities (Fig. 3A, B respectively). Furthermore, CADO at 20 μM significantly suppressed mRNA expression of TNAP (Fig. 3C), but not those of RUNX2 and BSP-II (Fig. 3C).


We first examined the expression of AR subtypes in HASMCs utilized in this study by RT-PCR. Messenger RNA of each of the four ARs was detected in HASMCs (Fig. 4A). To determine AR subtypes involved in the inhibitory effects in HASMC, the effects of various AR agonists on in vitro calcification and ALP activities were examined. CCPA (A1 AR-selective agonist), CGS21680 (A2a AR-selective agonist), and BAY60-6583 (A2b AR-selective agonist) failed to inhibit mineralization and ALP activities of HASMCs (Fig. 4C, D). On the other hand, IB-MECA (A3 AR-selective agonist) dose-dependently reduced both in vitro mineralization and ALP activities (Fig. 4C, D). A3 AR protein expression in HASMCs was not remarkably changed on day 4 by the treatment with various reagents (Fig. 4B). Furthermore, IB-MECA also significantly inhibited mRNA expression of TNAP, but not those of RUNX2 and BSP-II (Fig. 4E).
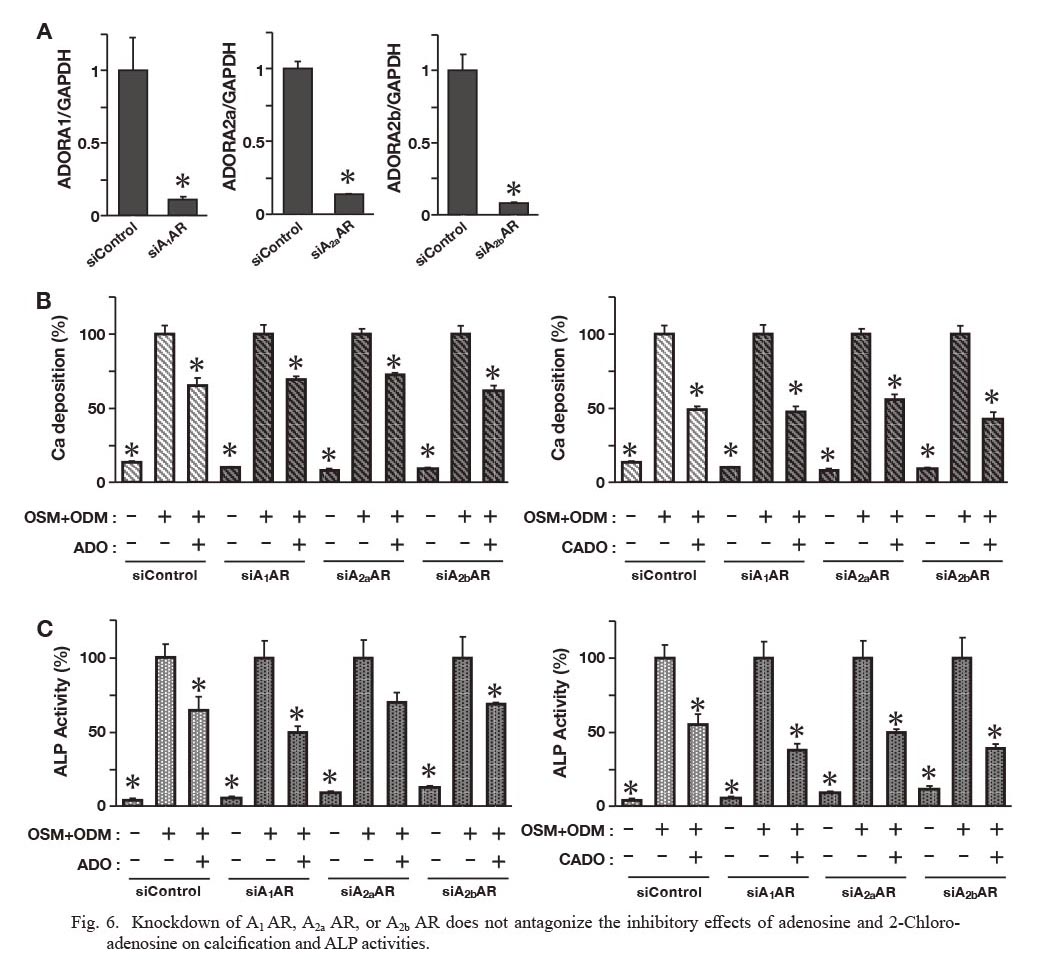
To further confirm the involvement of A3 AR in the inhibitory actions of adenosine signaling, we next investigated the effect of A3 AR knockdown using siRNA on the inhibitory action of IB-MECA on in vitro calcification and ALP activities of HASMC. Western blotting confirmed that treatment with siRNA silenced the expression of A3 AR in HASMCs (Fig. 5A). Knockdown of A3 AR significantly blocked the inhibitory actions of adenosine (100 μM), CADO (20 μM), and IB-MECA (100 nM) on mineralization of HASMCs and ALP activities (Fig. 5B, C). On the other hand, knockdown of other ARs such as A1 AR, A2a AR, and A2b AR, did not reverse the inhibitory actions of adenosine and CADO on in vitro calcification and ALP activities in HASMCs (Fig. 6A-C). These data suggest that A3 AR activation mediates the inhibitory effects of adenosine signaling on HASMC calcification.

Discussion
The current study clearly shows that adenosine attenuates vascular calcification in the culture model utilizing HASMCs. Adenosine potently reduces not only in vitro calcification, but also TNAP expression and its activities through A3 AR. To our knowledge, this is the first study to demonstrate the inhibitory effect of adenosine through A3 AR on HVSMC calcification in vitro.
Adenosine is generated from extracellular ATP by the action of the ectonucleotidases such as CD39 and CD73 (Antonioli et al. 2013). CD73 converts AMP to adenosine and inorganic phosphate. Arterial calcification observed in patients with CD73 mutations may be caused by attenuated inhibition of TNAP due to decreased levels of adenosine (St Hilaire et al. 2011). However, this putative function of adenosine to regulate vascular calcification through inhibition of TNAP expression has not so far been proved in the cultures of VSMC. Furthermore, the inhibitory action of adenosine on inflammation-induced TNAP expression in VSMC has not been clarified. In this study, we clearly demonstrated that adenosine inhibits OSM-induced calcification and TNAP expression in HASMCs. Since TNAP plays a pivotal role in not only skeletal mineralization but also vascular calcification (Shioi et al. 2002; Sheen et al. 2015), adenosine may exert its inhibitory action on vascular calcification through inhibiting TNAP expression in VSMCs. However, adenosine and its analogues did not down-regulate RUNX2 gene expression in HASMCs as shown in the present study. RUNX2 is a master regulator of osteoblast differentiation through up-regulating osteoblast-specific genes such as TNAP and BSP-II and its transcriptional activities are regulated by mitogen-activated protein kinase (MAPK)-dependent phosphorylation (Greenblatt et al. 2013). It is, therefore, likely that adenosine may regulate gene expression of TNAP and BSP-II through modulating RUNX2 phosphorylation by MAPK, but not its expression levels. Further studies are necessary to clarify the effects of adenosine on RUNX2 phosphorylation during OSM-induced osteoblastic differentiation of HASMC.
As shown in this study, adenosine exerted its inhibitory effects on HASMC calcification at concentrations between 10 and 100 μM. Because of the rapid cellular uptake and intracellular metabolism, the half-life of adenosine in the blood is counted in seconds (Riksen et al. 2008). Hence, the action of adenosine is restricted to the sites of tissue injury, inflammation, or hypoxia where the extracellular concentrations of adenosine can increase to about 30 μM from the baseline (20 to 300 nM) (Chen et al. 2013). Therefore, the doses of adenosine utilized in this study seem to be comparable to its tissue concentrations attainable under certain pathological conditions.
As also shown in this study, activation of A3 AR by its agonists such as adenosine, CADO, and IB-MECA inhibited TNAP expression and in vitro calcification in HASMCs, suggesting that adenosine may inhibit osteoblastic differentiation of VSMCs through A3 AR. It has been clarified that ARs play a pivotal role in osteoblast differentiation and function (Strazzulla and Cronstein 2016). Adenosine promotes osteogenic differentiation of mesenchymal stem cells mainly through A2b AR (Carroll et al. 2012; Takedachi et al. 2012). A2a AR also stimulates bone regeneration in a mouse critical-sized calvarial defect model (Mediero et al. 2015). A1 AR activation may enhance osteogenic differentiation of human dental pulp-derived stem cells through Wnt pathway (D’Alimonte et al. 2013). On the other hand, A3 AR stimulation has not been shown to have any effect on osteoblastic differentiation (Strazzulla and Cronstein 2016). Recently, it has been demonstrated that A2b AR activation reduces calcification in induced pluripotent stem cell-derived mesenchymal stromal cells and teratomas derived from patients with arterial calcification of CD73 deficiency (Jin et al. 2016). Additionally, in the culture of human valve interstitial cells (VICs), adenosine promoted in vitro mineralization through A2a AR, whereas A1 AR is involved in inhibition of human VIC calcification (Mahmut et al. 2015). However, there has been no report demonstrating the roles of ARs in regulating VSMC calcification. Therefore, it is likely that adenosine may specifically regulate vascular calcification in VSMC through A3 AR.
In conclusion, this study has reported that adenosine attenuates OSM-induced HASMC calcification through A3 AR. The A3 AR subtype in VSMCs may be a novel therapeutic target for atherosclerotic calcification.
Acknowledgments
This work was supported by JSPS KAKENHI Grant Number JP17K09731. We thank Asako Sawada for expert technical assistance.
Conflict of Interest
The authors declare no conflict of interest.
References
-
Antonioli,
L.,
Pacher,
P.,
Vizi,
E.S. &
Hasko,
G.
(2013) CD39 and CD73 in immunity and inflammation. Trends Mol. Med., 19, 355-367.
-
Benz,
K.,
Hilgers,
K.F.,
Daniel,
C. &
Amann,
K.
(2018) Vascular calcification in chronic kidney disease: the role of inflammation. Int. J. Nephrol., 2018, 4310379.
-
Borea,
P.A.,
Varani,
K.,
Vincenzi,
F.,
Baraldi,
P.G.,
Tabrizi,
M.A.,
Merighi,
S. &
Gessi,
S.
(2015) The A3 adenosine receptor: history and perspectives. Pharmacol. Rev., 67, 74-102.
-
Budoff,
M.J.,
Nasir,
K.,
Katz,
R.,
Takasu,
J.,
Carr,
J.J.,
Wong,
N.D.,
Allison,
M.,
Lima,
J.A.,
Detrano,
R.,
Blumenthal,
R.S. &
Kronmal,
R.
(2011) Thoracic aortic calcification and coronary heart disease events: the multi-ethnic study of atherosclerosis (MESA). Atherosclerosis, 215, 196-202.
-
Carroll,
S.H.,
Wigner,
N.A.,
Kulkarni,
N.,
Johnston-Cox,
H.,
Gerstenfeld,
L.C. &
Ravid,
K.
(2012) A2B adenosine receptor promotes mesenchymal stem cell differentiation to osteoblasts and bone formation in vivo. J. Biol. Chem., 287, 15718-15727.
-
Ceneri,
N.,
Zhao,
L.,
Young,
B.D.,
Healy,
A.,
Coskun,
S.,
Vasavada,
H.,
Yarovinsky,
T.O.,
Ike,
K.,
Pardi,
R.,
Qin,
L.,
Qin,
L.,
Tellides,
G.,
Hirschi,
K.,
Meadows,
J.,
Soufer,
R., et al.
(2017) Rac2 modulates atherosclerotic calcification by regulating macrophage interleukin-1beta production. Arterioscler. Thromb. Vasc. Biol., 37, 328-340.
-
Chen,
J.,
Budoff,
M.J.,
Reilly,
M.P.,
Yang,
W.,
Rosas,
S.E.,
Rahman,
M.,
Zhang,
X.,
Roy,
J.A.,
Lustigova,
E.,
Nessel,
L.,
Ford,
V.,
Raj,
D.,
Porter,
A.C.,
Soliman,
E.Z.,
Wright,
J.T. Jr., et al.
(2017) Coronary artery calcification and risk of cardiovascular disease and death among patients with chronic kidney disease. JAMA Cardiol., 2, 635-643.
-
Chen,
J.F.,
Eltzschig,
H.K. &
Fredholm,
B.B.
(2013) Adenosine receptors as drug targets: what are the challenges? Nat. Rev. Drug Discov., 12, 265-286.
-
D’Alimonte,
I.,
Nargi,
E.,
Lannutti,
A.,
Marchisio,
M.,
Pierdomenico,
L.,
Costanzo,
G.,
Di Iorio,
P.,
Ballerini,
P.,
Giuliani,
P.,
Caciagli,
F. &
Ciccarelli,
R.
(2013) Adenosine A1 receptor stimulation enhances osteogenic differentiation of human dental pulp-derived mesenchymal stem cells via WNT signaling. Stem Cell Res., 11, 611-624.
-
Demer,
L.L.
(1995) A skeleton in the atherosclerosis closet. Circulation, 92, 2029-2032.
-
Durham,
A.L.,
Speer,
M.Y.,
Scatena,
M.,
Giachelli,
C.M. &
Shanahan,
C.M.
(2018) Role of smooth muscle cells in vascular calcification: implications in atherosclerosis and arterial stiffness. Cardiovasc. Res., 114, 590-600.
-
Fish,
R.S.,
Klootwijk,
E.,
Tam,
F.W.,
Kleta,
R.,
Wheeler,
D.C.,
Unwin,
R.J. &
Norman,
J.
(2013) ATP and arterial calcification. Eur. J. Clin. Invest., 43, 405-412.
-
Greenblatt,
M.B.,
Shim,
J.H. &
Glimcher,
L.H.
(2013) Mitogen-activated protein kinase pathways in osteoblasts. Annu. Rev. Cell Dev. Biol., 29, 63-79.
-
Hortells,
L.,
Sur,
S. &
St Hilaire,
C.
(2018) Cell phenotype transitions in cardiovascular calcification. Front. Cardiovasc. Med., 5, 27.
-
Jin,
H.,
St Hilaire,
C.,
Huang,
Y.,
Yang,
D.,
Dmitrieva,
N.I.,
Negro,
A.,
Schwartzbeck,
R.,
Liu,
Y.,
Yu,
Z.,
Walts,
A.,
Davaine,
J.M.,
Lee,
D.Y.,
Donahue,
D.,
Hsu,
K.S.,
Chen,
J., et al.
(2016) Increased activity of TNAP compensates for reduced adenosine production and promotes ectopic calcification in the genetic disease ACDC. Sci. Signal., 9, ra121.
-
Kakutani,
Y.,
Shioi,
A.,
Shoji,
T.,
Okazaki,
H.,
Koyama,
H.,
Emoto,
M. &
Inaba,
M.
(2015) Oncostatin M promotes osteoblastic differentiation of human vascular smooth muscle cells through JAK3-STAT3 pathway. J. Cell. Biochem., 116, 1325-1333.
-
Kanda,
Y.
(2013) Investigation of the freely available easy-to-use software ‘EZR’ for medical statistics. Bone Marrow Transplant., 48, 452-458.
-
Kawada,
S.,
Nagasawa,
Y.,
Kawabe,
M.,
Ohyama,
H.,
Kida,
A.,
Kato-Kogoe,
N.,
Nanami,
M.,
Hasuike,
Y.,
Kuragano,
T.,
Kishimoto,
H.,
Nakasho,
K. &
Nakanishi,
T.
(2018) Iron-induced calcification in human aortic vascular smooth muscle cells through interleukin-24 (IL-24), with/without TNF-alpha. Sci. Rep., 8, 658.
-
Leopold,
J.A.
(2015) Vascular calcification: mechanisms of vascular smooth muscle cell calcification. Trends Cardiovasc. Med., 25, 267-274.
-
Mahmut,
A.,
Boulanger,
M.C.,
Bouchareb,
R.,
Hadji,
F. &
Mathieu,
P.
(2015) Adenosine derived from ecto-nucleotidases in calcific aortic valve disease promotes mineralization through A2a adenosine receptor. Cardiovasc. Res., 106, 109-120.
-
Mediero,
A.,
Wilder,
T.,
Perez-Aso,
M. &
Cronstein,
B.N.
(2015) Direct or indirect stimulation of adenosine A2A receptors enhances bone regeneration as well as bone morphogenetic protein-2. FASEB J., 29, 1577-1590.
-
Riksen,
N.P.,
Rongen,
G.A.,
Yellon,
D. &
Smits,
P.
(2008) Human in vivo research on the vascular effects of adenosine. Eur. J. Pharmacol., 585, 220-227.
-
Santos,
R.D.,
Rumberger,
J.A.,
Budoff,
M.J.,
Shaw,
L.J.,
Orakzai,
S.H.,
Berman,
D.,
Raggi,
P.,
Blumenthal,
R.S. &
Nasir,
K.
(2010) Thoracic aorta calcification detected by electron beam tomography predicts all-cause mortality. Atherosclerosis, 209, 131-135.
-
Sheen,
C.R.,
Kuss,
P.,
Narisawa,
S.,
Yadav,
M.C.,
Nigro,
J.,
Wang,
W.,
Chhea,
T.N.,
Sergienko,
E.A.,
Kapoor,
K.,
Jackson,
M.R.,
Hoylaerts,
M.F.,
Pinkerton,
A.B.,
O’Neill,
W.C. &
Millan,
J.L.
(2015) Pathophysiological role of vascular smooth muscle alkaline phosphatase in medial artery calcification. J. Bone Miner. Res., 30, 824-836.
-
Shioi,
A. &
Ikari,
Y.
(2018) Plaque calcification during atherosclerosis progression and regression. J. Atheroscler. Thromb., 25, 294-303.
-
Shioi,
A.,
Katagi,
M.,
Okuno,
Y.,
Mori,
K.,
Jono,
S.,
Koyama,
H. &
Nishizawa,
Y.
(2002) Induction of bone-type alkaline phosphatase in human vascular smooth muscle cells: roles of tumor necrosis factor-alpha and oncostatin M derived from macrophages. Circ. Res., 91, 9-16.
-
St Hilaire,
C.,
Ziegler,
S.G.,
Markello,
T.C.,
Brusco,
A.,
Groden,
C.,
Gill,
F.,
Carlson-Donohoe,
H.,
Lederman,
R.J.,
Chen,
M.Y.,
Yang,
D.,
Siegenthaler,
M.P.,
Arduino,
C.,
Mancini,
C.,
Freudenthal,
B.,
Stanescu,
H.C., et al.
(2011) NT5E mutations and arterial calcifications. N. Engl. J. Med., 364, 432-442.
-
Strazzulla,
L.C. &
Cronstein,
B.N.
(2016) Regulation of bone and cartilage by adenosine signaling. Purinergic Signal., 12, 583-593.
-
Takedachi,
M.,
Oohara,
H.,
Smith,
B.J.,
Iyama,
M.,
Kobashi,
M.,
Maeda,
K.,
Long,
C.L.,
Humphrey,
M.B.,
Stoecker,
B.J.,
Toyosawa,
S.,
Thompson,
L.F. &
Murakami,
S.
(2012) CD73-generated adenosine promotes osteoblast differentiation. J. Cell. Physiol., 227, 2622-2631.
-
Tintut,
Y.,
Patel,
J.,
Parhami,
F. &
Demer,
L.L.
(2000) Tumor necrosis factor-alpha promotes in vitro calcification of vascular cells via the cAMP pathway. Circulation, 102, 2636-2642.
-
Wilson,
P.W.,
Kauppila,
L.I.,
O’Donnell,
C.J.,
Kiel,
D.P.,
Hannan,
M.,
Polak,
J.M. &
Cupples,
L.A.
(2001) Abdominal aortic calcific deposits are an important predictor of vascular morbidity and mortality. Circulation, 103, 1529-1534.