Abstract
We herein present the case of a 45-year-old diabetic woman who developed diabetic ketoacidosis following the administration of dapagliflozin, a sodium-glucose cotransporter 2 (SGLT2) inhibitor. The patient had been diagnosed with diabetes three years previously and was being treated with multiple daily injections of insulin. Metformin hydrochloride and dapagliflozin were added seven months and 11 months later, respectively. Her clinical course was uneventful until the onset of influenza. She then discontinued insulin and oral medications voluntarily. On arrival at the hospital, she was found to be in a state of ketoacidosis, and promptly received insulin and saline infusion. In retrospect, the initial amount of glucose infused was insufficient, and the hypoglycemia was thought to have been prolonged. This phenomenon may also have affected her long-term urinary glucose excretion. Her urinary L-type fatty acid-binding protein (L-FABP) level was found to be markedly elevated (48.8 μg/g·Cr, reference value < 8.4 μg/g·Cr) as was her urinary β2-microglobulin level (9,230 μg/L, reference value < 230 μg/L). Patients with SGLT-2 inhibitor-associated diabetic ketoacidosis often exhibit protracted hyperglycosuria, in which acute proximal renal tubular dysfunction is considered to be etiologically implicated.
Introduction
Diabetic ketoacidosis (DKA) may occur as an adverse event in patients treated with sodium-glucose cotransporter 2 (SGLT2) inhibitors. This phenomenon has been described in numerous case reports (Bonora et al. 2018; Hampp et al. 2020). The following four points are important in the treatment of SGLT2 inhibitor-associated DKA: (1) Stopping the SGLT2 inhibitor, (2) increasing the insulin dose, (3) providing adequate carbohydrate replacement, and (4) providing adequate fluids (Garg et al. 2018; Danne et al. 2019). Some case reports of SGLT2 inhibitor-associated DKA have described rapid improvement in symptoms (Fukuyama et al. 2020), while others have described persistent acidosis, prolonged positive urine glucose, and osmotic diuresis (Adachi et al. 2017; Kelmenson et al. 2017; Miyauchi et al. 2017; Pujara and Ioachimescu 2017; Wang and Isom 2020). We experienced a case of SGLT2 inhibitor-associated DKA that resulted in prolonged urinary glucose excretion even during persistent hypoglycemia. Retrospectively, we consider that the glucose supplementation, in the present case, was not sufficient as an initial treatment. During the prolonged hypoglycemia, the patient’s urine was strongly positive for glucose and the excretion of glucose in the urine was prolonged. This patient had elevated low molecular weight protein, urinary β2-microglobulin (β2MG), and urinary L-type fatty acid-binding protein (L-FABP), and acute proximal tubular dysfunction was suspected. We report this case because the prolonged urinary glucose excretion in DKA with SGLT2 inhibitor is thought to have been due to the persistent effect of the drug, but acute proximal tubular dysfunction may have also been involved.
Case Presentation
The patient was a 45-year-old woman who had been diagnosed with diabetes three years ago. At her first visit, her hemoglobin A1c (HbA1c) was 7.8% (61 mmol/mol), her fasting blood glucose was 180 mg/dl, her C-peptide index (CPI) was 1.1, and a test for glutamic acid decarboxylase (GAD) antibody was negative. The patient was diagnosed with type 2 diabetes and started on a diet. Due to her poor glycemic control, intensive insulin therapy was started. In addition, she was managed with metformin hydrochloride (500 mg) seven months later and dapagliflozin (5 mg) after 11 months later. The patient’s fasting C-peptide immunoreactivity (CPR) was 0.39 ng/ml, her fasting blood glucose level was 165 mg/dl, and her insulin secretion capacity had been declining in the three months before admission (Table 1).
Four days before admission, she developed fever, vomiting, nausea, and anorexia. Thus, she discontinued her insulin injections and oral medications at her discretion. She tested positive for influenza A antigen three days before admission. She took laninamivir octanoate hydrate and acetaminophen. She quickly recovered from the fever after taking anti-influenza medication. However, due to persistent nausea and loss of appetite, she visited our emergency department on the day before admission and received infusion therapy. The doctor instructed her to resume insulin glargine. As her symptoms did not improve on the day of admission, she returned the hospital. A blood test revealed acidosis, and she was admitted to the hospital to begin treatment.
On examination, the patient’s blood pressure was 130/94 mmHg, her heart rate was 104 beats/min, and her temperature was 36.9℃. Her weight, height, and body mass index were 43.8 kg, 152 cm, and 18.96 kg/m2, respectively. Her oral mucosa was dry. The results of other neurological and general examinations were normal. Dehydration was suspected based on the results of blood tests on admission. The patient was positive for urine ketone bodies, her blood 3-hydroxybutyrate value was 8,085μM/L, and venous blood gases showed marked acidosis at pH 7.071 and HCO3− 6.6 mmol/L. Her HbA1c level was 7.3% (56 mmol/mol), and her blood glucose level was 371 mg/dl (Table 2).
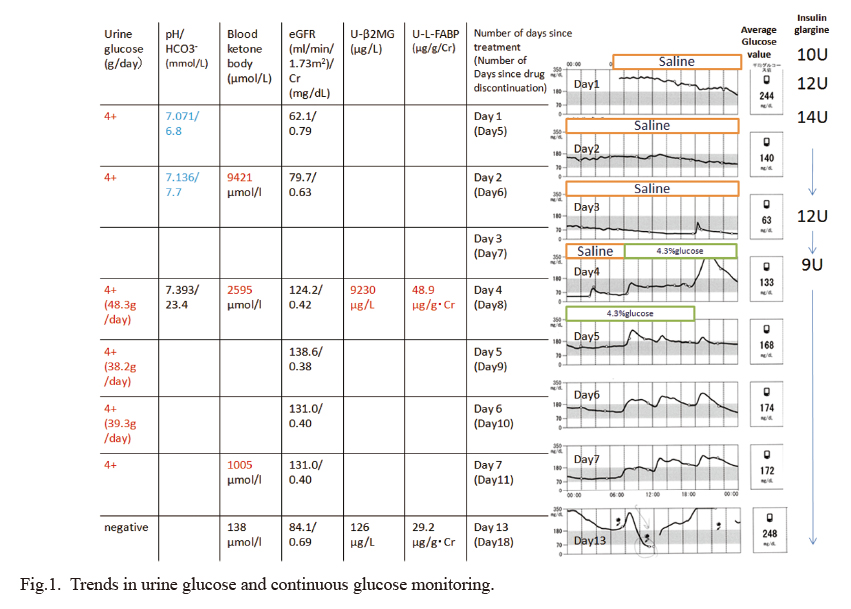
She was initially treated with massive isotonic saline. Although she had acidosis, her serum glucose level was moderately elevated. We determined that she would be able to take food orally if we treated her for dehydration. For this reason, continuous insulin infusion was not started. Her dose of insulin glargine was then increased from the usual 10 units to 12 units. We were going to give her subcutaneous injections of insulin aspart depending on her blood glucose levels. On the second day of hospitalization, her hyperglycemia was improving, however, her acidosis persisted. We decided that the insulin was insufficient to inhibit fat beta-oxidation and increased the insulin glargine dose to 14 units. From the third day of hospitalization, she began to have prolonged hypoglycemia. She also had persistent nausea and loss of appetite. On the fourth day of hospitalization, believing that the glucose replacement was inadequate, she was started on a 4.3% glucose-containing infusion. Subsequently, her nausea promptly disappeared, and her appetite improved. Thereafter, the preprandial administration of insulin aspart was restarted. Her general condition improved and she was discharged on the eighth day of hospitalization. Her glycosuria remained high until the eighth day of hospitalization. At an outpatient visit after her discharge from the hospital (day 13 of admission), a urinary glucose test was negative (Fig. 1).
On the 3rd day after admission, even though she had hypoglycemia, a urine test glucose was strongly positive. Therefore, we suspected that her renal tubules were not functioning. In addition to impaired glucose reabsorption, there was an increase in urinary β2MG (9,230μg/L, reference value < 230μg/L) and urinary L-FABP (48.8μg/g·Cr, reference value < 8.4μg/g·Cr) suggesting the presence of acute proximal renal tubular dysfunction. Her urinary β2MG and urinary L-FABP were confirmed to be normal in the outpatient clinic after discharge (13th day of hospitalization). On the seventh day of hospitalization, her urinary CPR was as low as 5.6μg/day. Tests for GAD and insulinoma-associated antigen-2 (IA-2) antibodies were negative. Her pancreas-related autoantibodies were negative, but her insulin secretion capacity decreased. She was eventually diagnosed with idiopathic type 1 diabetes mellitus (Table 1).
Discussion
There have been cases of SGLT2 inhibitor-associated DKA in which urinary glucose excretion persists for 4-5 days after the discontinuation of SGLT2 inhibitors (Adachi et al. 2017; Kelmenson et al. 2017; Miyauchi et al. 2017; Pujara and Ioachimescu 2017; Wang and Isom 2020). There are four possible mechanisms by which urinary glucose excretion may be prolonged. (1) Blood concentrations of SGLT2 inhibitors remain relatively high even after the drug is discontinued (Adachi et al. 2017). (2) Prolonged acidosis prolongs protein binding to SGLT2 inhibitors, and when acidosis improves, SGLT2 inhibitors dissociate from serum proteins, exert their SGLT2 inhibition, and prolong urinary glucose excretion (Miyauchi et al. 2017). (3) The administration of SGLT2 inhibitors decreases not only the expression of SGLT2 but also the expression of SGLT1 in the proximal tubules, resulting in prolonged urinary glucose excretion, even after the discontinuation of SGLT2 inhibitor (Miyauchi et al. 2017). (4) Single Nucleotide Polymorphism (SNP) mutations in uridine diphosphate glucuronosyltransferase (UGT), the drug-metabolizing enzyme of SGLT2 inhibitors, are associated with a delayed drug metabolism (Kelmenson et al. 2017). In this case, (1), (2), and (3) could not be evaluated because we were not able to measure the blood and urine concentrations of dapagliflozin in order to examine the expression of SGLT1/2. The metabolic enzyme for dapagliflozin is UGT1A (Kasichayanula et al. 2014). The involvement of UGT1A9 SNP mutation in (4) is a possibility; however, we were not able to evaluate this in the present case.
The patient had prolonged hypoglycemia with a mean blood glucose level of 63 mg/dl on continuous glucose monitoring on the third day of admission. And an early morning urinalysis on her fourth day of hospitalization showed urine glucose 4+ (urine glucose 48.3 g/day) (Fig. 1). We considered the following four possible explanations of the prolonged hypoglycemia in this patient. (1) If both SGLT1 and 2 were functioning: urine glucose would be negative, assuming a tubular glucose reabsorption threshold of about 160 mg/dl in this case. (2) If SGLT2 was functional and only SGLT1 was impaired: the patient would be negative for urine glucose, considering that SGLT2 is responsible for 90% of urine glucose reabsorption (Abdul-Ghani et al. 2013). (3) If SGLT1 was functioning and only SGLT2 was impaired: the threshold for tubular glucose reabsorption is 37 mg/dl in healthy subjects and 21 mg/dl in patients with type 2 diabetes when taking dapagliflozin, and assuming that this patient has a glomerular filtration rate (GFR) of 80 ml/min, the daily excretion of urinary glucose would be approximately 25-50 g (DeFronzo et al. 2013). (4) If both SGLT1 and SGLT2 were not functioning: the glucose reabsorption threshold of the renal tubules would be zero, and the daily urinary glucose excretion would be approximately 70 g. We did not measure the daily urinary glucose excretion during prolonged hypoglycemia in this patient. It is possible that either only SGLT2 or both SGLT1 and SGLT2 were not functioning in this patient even on day 7 after discontinuing dapagliflozin medication (Table 3).
Recently, cases of acute kidney injury (AKI) in patients on SGLT2 inhibitor medication have been reported, and the U.S. Food and Drug Administration Drug Safety Communication issued a cautionary warning in 2016 (U.S. Food and Drug Administration 2016). The mechanism of SGLT2 inhibitor-induced AKI has been discussed as follows. (1) In the diabetic kidney, the amount of glucose that reaches the proximal tubules is high. Therefore, the workload of SGLT2 is higher, the partial pressure of oxygen at the renal corticomedullary border is lower than in normal kidneys (Palm et al. 2003; Heyman et al. 2013). When SGLT2 inhibitors are administered to diabetic animal models, the workload of SGLT2 is reduced, resulting in improved partial pressure of oxygen in the renal cortex. However, in the proximal tubular region of the S3 segment, where SGLT1 is distributed, the amount of glucose reaching the tubules increases, followed by an increase in workload, which results in a decrease in the partial pressure of oxygen in the renal medulla (O’Neill et al. 2015). It has been reported that the increase in erythropoietin and reticulocyte levels in patients treated with SGLT2 inhibitors is due to the renoprotective effect of SGLT2 inhibitors (Sano et al. 2016). However, some believe that a decrease in the partial pressure of oxygen at the renal corticomedullary border activates hypoxia-inducible factors (HIFs), increases erythropoietin production, and increases the number of reticulocytes (Heyman et al. 2017b). Therefore, when SGLT2 inhibitors are used in combination with conditions or drugs that induce hypoxia in the renal medulla, such as decreased fluid volume, nonsteroidal anti-inflammatory drugs (NSAIDs), angiotensin receptor blockers (ARBs), angiotensin-converting enzyme (ACE) inhibitors, and radiocontrast agents, it is necessary to pay close attention to the development of AKI (Heyman et al. 2017a). (2) It is thought to be related to increased renal uric acid excretion. Glucose is exchanged for uric acid by glucose transporter (GLUT)-9, glucose in urine is reabsorbed, and uric acid is excreted. The administration of SGLT2 inhibitors increases urinary glucose in the tubules of the S3 segment region, so that the exchange transport works and uric acid excretion increases. Increased uric acid excretion may lead to uric acid crystal formation and direct tubular damage (Hahn et al. 2016). (3) The involvement of increased fructose metabolism has been considered. Increased urinary glucose reabsorption in the tubules of the S3 segment region activates aldose reductase and increases fructose production. Fructokinase is highly expressed in the tubules of the S3 segment, and metabolized fructose may cause uric acid production, oxidative stress, and chemokine release, leading to local tubular damage and inflammation (Hahn et al. 2016). In the cases that developed AKI while taking SGLT2 inhibitors and in which renal biopsy was performed, findings of osmotic nephropathy and acute tubular necrosis were observed; histologically, tubular injury was the main pathology (Pleros et al. 2018; Phadke et al. 2020). In a study evaluating the real-world risk of AKI in new users of SGLT2 inhibitors in a large health care utilization cohort of patients with type 2 diabetes, the use of SGLT2 inhibitors did not increase the risk of developing AKI (Menne et al. 2019), and in a meta-analysis examining the effect of SGLT2 inhibitors on renal adverse events, SGLT2 inhibitors reduced the risk of AKI (Nadkarni et al. 2017). The potential risk of AKI due to SGLT2 inhibitors occurs in a population with a high risk of renal impairment; thus, nephrotoxicity is not likely to occur if SGLT2 inhibitors are used appropriately. However, we speculate that SGLT2 inhibitors may cause tubular damage if not used properly.
The proposed pathogenesis of the present case is as follows. The patient had decreased insulin secretion capacity and was started on insulin therapy. However, her glycemic control was poor and she was started on SGLT2 inhibitor treatment. Reabsorption of glucose via SGLT2 in the S1/2 region of the proximal tubule was inhibited, urinary glucose excretion was increased, and glycemic control was improved. However, on the other hand, glucose reabsorption of SGLT1 in the S3 region of the proximal tubule was enhanced, and adenosine triphosphate (ATP) and oxygen consumption in the border region of the renal cortex medulla may have been increased. In this situation, the patient had influenza and developed SGLT2 inhibitor-associated DKA triggered by insulin interruption. Due to influenza, her carbohydrate intake was decreased, renal glycogenesis was increased, and her renal ATP and oxygen requirements may have increased. Furthermore, due to the absolute lack of insulin, glucose as a substrate for the production of ATP became unavailable, and lipolysis and ketone body production increased. From this point, treatment was initiated, insulin was supplemented, and glucose became available as an energy substrate. However, the fasting period was long, her liver glycogen was thought to have been depleted, and the glucose replacement was inadequate. Therefore, it is predicted that the ATP supply was insufficient and that glycogenesis did not work properly. The prolonged excretion of urinary glucose by SGLT2 inhibitor-associated DKA may be related to the impairment of the SGLT1 and SGLT2 functions due to acute proximal tubular injury caused by energy substrate deficiency. In fact, in this case, urine glucose tests were strongly positive and urinary β2MG and L-FABP were elevated during the period of prolonged urinary glucose positivity on the third day of hospitalization, suggesting that acute proximal tubular damage may have occurred.
One of the problems, in the present case, is that the patient did not receive continuous insulin infusion during treatment of DKA. The blood concentration of insulin required to express 50% inhibition of lipolysis in adipocytes is 7-17μU/ml, and even in the state of DKA, basal secretion of 50μU/ml is sufficient to inhibit lipolysis (Stumvoll and Jacob 1999). In this case, we were not able to measure the patient’s blood insulin levels while she was receiving treatment for DKA. However, subcutaneous injection of insulin glargine was started, and acidosis improved at 60 hours after admission. We therefore believe that the insulin dose was sufficient to suppress lipolysis.
We experienced a case of SGLT2 inhibitor-associated DKA after influenza infection and the interruption of insulin therapy in a diabetic patient with impaired insulin secretion. In this case, despite the prolonged hypoglycemia on the third day of treatment (seventh day of drug discontinuation), the patient was strongly positive for urinary glucose. We therefore considered that tubular damage was present. It is thought that the workload of SGLT1 is increased during SGLT2 inhibitor treatment, and that the ATP and oxygen requirements of the renal corticomedullary border are increased. We hypothesized that if DKA develops in this state and glucose supplementation becomes inadequate, acute damage to the proximal tubules will occur due to insufficient ATP supply, leading to a decreased SGLT2/1 function and prolonged urinary glucose excretion. As has been mentioned, adequate glucose replacement is important in the treatment of SGLT2 inhibitor-associated DKA.
Conflict of Interest
The authors declare no conflict of interest.
References
-
Abdul-Ghani,
M.A.,
DeFronzo,
R.A. &
Norton,
L.
(2013) Novel hypothesis to explain why SGLT2 inhibitors inhibit only 30-50% of filtered glucose load in humans. Diabetes, 62, 3324-3328.
-
Adachi,
J.,
Inaba,
Y. &
Maki,
C.
(2017) Euglycemic diabetic ketoacidosis with persistent diuresis treated with canagliflozin. Intern. Med., 56, 187-190.
-
Bonora,
B.M.,
Avogaro,
A. &
Fadini,
G.P.
(2018) Sodium-glucoseco-transporter-2 inhibitors and diabetic ketoacidosis: an updated review of the literature. Diabetes Obes. Metab., 20, 25-33.
-
Danne,
T.,
Garg,
S.,
Peters,
A.L.,
Buse,
J.B.,
Mathieu,
C.,
Pettus,
J.H.,
Alexander,
C.M.,
Battelino,
T.,
Ampudia-Blasco,
F.J.,
Bode,
B.W.,
Cariou,
B.,
Close,
K.L.,
Dandona,
P.,
Dutta,
S.,
Ferrannini,
E.,
et al. (2019) International consensus on risk management of diabetic ketoacidosis in patients with type 1 diabetes treated with sodium-glucose cotransporter (SGLT) inhibitors. Diabetes Care, 42, 1147-1154.
-
DeFronzo,
R.A.,
Hompesch,
M.,
Kasichayanula,
S.,
Liu,
X.,
Hong,
Y.,
Pfister,
M.,
Morrow,
L.A.,
Leslie,
B.R.,
Boulton,
D.W.,
Ching,
A.,
LaCreta,
F.P. &
Griffen,
S.C.
(2013) Characterization of renal glucose reabsorption in response to dapagliflozin in healthy subjects and subjects with type 2 diabetes. Diabetes Care, 36, 3169-3176.
-
Fukuyama,
Y.,
Numata,
K.,
Yoshino,
K.,
Santanda,
T. &
Funakoshi,
H.
(2020) Euglycemic diabetic ketoacidosis due to a strict low-carbohydrate diet during treatment with sodium-glucose cotransporter 2 inhibitors. Acute Med. Surg., 7, e480.
-
Garg,
S.K.,
Peters,
A.L.,
Buse,
J.B. &
Danne,
T.
(2018) Strategy for mitigating DKA risk in patients with type 1 diabetes on adjunctive treatment with SGLT inhibitors: a STICH Protocol. Diabetes Technol. Ther., 20, 571-575.
-
Hahn,
K.,
Ejaz,
A.A.,
Kanbay,
M.,
Lanaspa,
M.A. &
Johnson,
R.J.
(2016) Acute kidney injury from SGLT2 inhibitors: potential mechanisms. Nat. Rev. Nephrol., 12, 711-712.
-
Hampp,
C.,
Swain,
R.S.,
Horgan,
C.,
Dee,
E.,
Qiang,
Y.,
Dutcher,
S.K.,
Petrone,
A.,
Tilney,
R.C.,
Maro,
J.C. &
Panozzo,
C.A.
(2020) Use of sodium-glucose cotransporter 2 inhibitors in patients with type 1 diabetes and rates of diabetic ketoacidosis. Diabetes Care, 43, 90-97.
-
Heyman,
S.N.,
Khamaisi,
M.,
Rosen,
S.,
Rosenberger,
C. &
Abassi,
Z.
(2017a) Potential hypoxic renal injury in patients with diabetes on SGLT2 inhibitors: caution regarding concomitant use of NSAIDs and iodinated contrast media. Diabetes Care, 40, e40-e41.
-
Heyman,
S.N.,
Khamaisi,
M.,
Rosenberger,
C.,
Szalat,
A. &
Abassi,
Z.
(2017b) Increased hematocrit during sodium-glucose cotransporter-2 inhibitor therapy. J. Clin. Med. Res., 9, 176-177.
-
Heyman,
S.N.,
Rosenberger,
C.,
Rosen,
S. &
Khamaisi,
M.
(2013) Why is diabetes mellitus a risk factor for contrast-induced nephropathy? BioMed Res. Int., 2013, 123589.
-
Kasichayanula,
S.,
Liu,
X.,
Lacreta,
F.,
Griffen,
S.C. &
Boulton,
D.W.
(2014) Clinical pharmacokinetics and pharmacodynamics of dapagliflozin, a selective inhibitor of sodium-glucose co-transporter type 2. Clin. Pharmacokinet., 53,17-27.
-
Kelmenson,
D.A.,
Burr,
K.,
Azhar,
Y.,
Reynolds,
P.,
Baker,
C.A. &
Rasouli,
N.
(2017) Euglycemic diabetic ketoacidosis with prolonged glucosuria associated with the sodium-glucose cotransporter-2 canagliflozin. J. Investig. Med. High Impact Case Rep., 5, 2324709617712736.
-
Menne,
J.,
Dumann,
E.,
Haller,
H. &
Schmidt,
B.M.W.
(2019) Acute kidney injury and adverse renal events in patients receiving SGLT2-inhibitors: a systematic review and meta-analysis. PLoS Med., 16, e1002983.
-
Miyauchi,
M.,
Toyoda,
M. &
Fukagawa,
M.
(2017) Atypical ketoacidosis and protracted hyperglycosuria after treatment with ipragliflozin, an SGLT2 inhibitor. Intern. Med., 56, 1673-1678.
-
Nadkarni,
G.N.,
Ferrandino,
R.,
Chang,
A.,
Surapaneni,
A.,
Chauhan,
K.,
Poojary,
P.,
Saha,
A.,
Ferket,
B.,
Grams,
M.E. &
Coca,
S.G.
(2017) Acute kidney injury in patients on SGLT2 inhibitors: a propensity-matched analysis. Diabetes Care, 40, 1479-1485.
-
O’Neill,
J.,
Fasching,
A.,
Pihl,
L.,
Patinha,
D.,
Franzen,
S. &
Palm,
F.
(2015) Acute SGLT inhibition normalizes O2 tension in the renal cortex but causes hypoxia in the renal medulla in anaesthetized control and diabetic rats. Am. J. Physiol. Renal Physiol., 309, F227-234.
-
Palm,
F.,
Cederberg,
J.,
Hansell,
P.,
Liss,
P. &
Carlsson,
P.O.
(2003) Reactive oxygen species cause diabetes-induced decrease in renal oxygen tension. Diabetologia, 46, 1153-1160.
-
Phadke,
G.,
Kaushal,
A.,
Tolan,
D.R.,
Hahn,
K.,
Jensen,
T.,
Bjornstad,
P.,
Roncal-Jimenez,
C.,
Hernando,
A.A.,
Lanaspa,
M.A.,
Alexander,
M.P.,
Kukla,
A. &
Johnson,
R.J.
(2020) Osmotic nephrosis and acute kidney injury associated with SGLT2 inhibitor use: a case report. Am. J. Kidney Dis., 76, 144-147.
-
Pleros,
C.,
Stamataki,
E.,
Papadaki,
A.,
Damianakis,
N.,
Poulidaki,
R.,
Gakiopoulou,
C. &
Tzanakis,
I.
(2018) Dapagliflozin as a cause of acute tubular necrosis with heavy consequences: a case report. CEN Case Rep., 7, 17-20.
-
Pujara,
S. &
Ioachimescu,
A.
(2017) Prolonged ketosis in a patient with euglycemic diabetic ketoacidosis secondary to dapagliflozin. J. Investig. Med. High Impact Case Rep., 5, 2324709617710040.
-
Sano,
M.,
Takei,
M.,
Shiraishi,
Y. &
Suzuki,
Y.
(2016) Increased hematocrit during sodium-glucose cotransporter 2 inhibitor therapy indicates recovery of tubulointerstitial function in diabetic kidneys. J. Clin. Med. Res., 8, 844-847.
-
Stumvoll,
M. &
Jacob,
S.
(1999) Multiple sites of insulin resistance: muscle, liver and adipose tissue. Exp. Clin. Endocrinol. Diabetes, 107, 107-110.
-
U.S. Food and Drug Administration
(2016) FDA Drug Safety Communication: FDA strengthens kidney warnings for diabetes medicines canagliflozin (Invokana, Invokamet) and dapagliflozin (Farxiga, XigduoXR). http://www.fda.gov/Drugs/DrugSafety/ucm505860.htm [Accessed: July 14, 2016].
-
Wang,
K.M. &
Isom,
R.T.
(2020) SGLT2 inhibitor-induced euglycemic diabetic ketoacidosis: a case report. Kidney Med., 2, 218-221.