Abstract
Esophageal squamous cell carcinoma (ESCC) showed limited treatment outcome and poor prognosis. This study aimed to screen potential biomarkers and drugs in ESCC. Firstly, GSE26886, GSE111044 and GSE77861 were downloaded from the Gene Expression Omnibus (GEO) database. Next, the differentially expressed genes (DEGs) between cancer and noncancerous tissues were analyzed by the GEO2R. The Gene Ontology (GO) annotation, the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway annotation, the protein-protein interaction (PPI) analysis and hub genes screened were conducted by some bioinformatic methods, respectively. Lastly, the hub genes and potential drugs were verified by the GEPIA2 and the QuartataWeb database. The results showed that 13 up-regulated genes and 81 down-regulated genes were identified. In GO terms, DEGs were mainly associated with cell proliferation, cell migration and cell differentiation. DEGs did not cluster into the KEGG pathway. After hub genes validated, nine genes (FLG, COL1A1, COL1A2, PSCA, SCEL, PPL, ACPP, CNFN, and A2ML1) expression trends showed no change. Moreover, higher COL1A1 or COL1A2 expression for ESCC patients showed poor prognosis. Finally, five drugs used for promoting blood coagulation were identified. Probably, these drugs could show anticancer effects by promoting blood coagulation or inhibiting vascular formation in cancers, which offers a novel idea for the treatment of ESCC.
Introduction
Esophageal cancer (EC) is the eighth common harmful cancer of digestive system, and is associated with a high mortality rate globally (Arnold et al. 2015). Esophageal squamous cell carcinoma (ESCC) is one of the sub-types of EC, which has been defined according to its epidemiology and histopathology (Forde and Kelly 2013). ESCC remains responsible for the most majority of EC cases (Liu et al. 2021), though its incidence is declining in most countries.
The prognosis of ESCC is poor, owing to the lack of effective early biomarkers and treatment strategies (Zhang and Jain 2020; Liu et al. 2021; Yang et al. 2021). The overall 5-year survival are less than 25% in the late-stage of ESCC patients. By contrast, the 5-year survival rates are more than 85% for patients with ESCC diagnosed in early stages (Lordick et al. 2016). The endoscopy is often used as a primary diagnostic tool to screen ESCC; however, its application is limited, owing to its serious side effects and dependence on the skill of the endoscopist (Marginean and Dhanpat 2020; Su et al. 2020; Zhang and Jain 2020). Moreover, due to its invasive nature, some patients refuse to undergo endoscopy (Lin et al. 2013). Therefore, identification of effective methods for the diagnosis of ESCC is imperative.
High-throughput sequencing has been broadly applied in many diseases, particularly, in cancers (Freimanis and Oade 2020; Kamio et al. 2020; Pillay et al. 2020). Various public databases are available for obtaining clinical and sequencing data that are uploaded after analysis. Reanalysis of these data through bioinformatic methods can provide new insights into cancers. Although some bioinformatic studies on ESCC have been conducted (Zhang et al. 2020; Xue et al. 2021; Ye et al. 2021), all these studies attempted to mine a single gene signature related to cancers, but a single gene may not be sufficient to understand the cancer, because tumorigenesis is a multi-factor, multi-stage, and multi-gene process. Moreover, these studies inadvertently ignored the genes and targeted drugs. Thus, the diagnosis of ESCC remains challenging, resulting in poor prognosis of patients.
“The idea of repurposing of old drugs” has received considerable attention of researchers who are aiming to develop an effective cancer therapy. Old drugs are defined as the Food and Drug Administration (FDA) approved drugs or those with safe clinical application. For example, metformin, a widely prescribed antidiabetic drug used to treat type II diabetes, has shown tumor suppressive properties (Quinn et al. 2013; Yue et al. 2014; Anisimov 2016). Interestingly, the combination of metformin with targeted drugs has been reported to improve targeted therapeutic efficacies in cancers (Morales and Morris 2015; Armstrong et al. 2021; Cunha Junior et al. 2021; Jang et al. 2021). Therefore, we propose the hypothesis whether the old drugs can be screened through bioinformatics methods to identify new anticancer drugs for ESCC.
In this study, comprehensive bioinformatic analyses were used to discover potential biomarkers and available drugs for ESCC. Firstly, three microarray datasets in the Gene Expression Omnibus (GEO) database were selected and analyzed. Then, the differentially expressed genes (DEGs) between ESCC and healthy groups were identified. Furthermore, the Gene Ontology (GO) annotation, the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway annotation, and protein protein interaction (PPI) analysis were conducted among the DEGs through several bioinformatics methods. Finally, the potential biomarkers and correlated pathways, as well as the drugs, which might be associated with ESCC, were identified. The candidate genes/drugs identified in the study may serve as promising diagnostic biomarkers or therapeutic targets for ESCC.
Materials and Methods
Microarray data information
GEO database possess lots of gene expression data, including high-throughput data, hybridization arrays and microarrays data (Barrett et al. 2013). GSE26886, GSE111044 and GSE77861 were downloaded from GEO database. The platforms of the microarray datasets were GPL570 (Affymetrix Human Genome U133 Plus 2.0 Array). GSE26886 comprised 21 cancer tissue samples and 9 normal tissue samples. GSE111044 comprised 3 cancer tissue samples and 3 normal tissue samples. GSE77861 comprised 7 cancer tissue samples and 7 normal tissue samples (Table 1).
Identification of DEGs
The DEGs between ESCC and normal specimen were found via the GEO2R. The cut‐off criteria were set as |logFC| > 2 and P value < 0.05. Then, the commonly DEGs among the three datasets were screened in Venn software. The logFC < −2 represented down-regulated genes, meanwhile logFC > 2 represented up-regulated genes among these DEGs.
Gene ontology analysis and KEGG pathway enrichment analysis
The Gene ontology (GO) analysis is used to identify the biological properties of genome data of genes or high-throughput transcriptome (Dalmer and Clugston 2019). The KEGG stores various databases, including genomes, biological pathways, diseases and drugs (Kanehisa et al. 2017). The Enrichr website (Kuleshov et al. 2016) was used to show the GO analysis and KEGG pathways analysis (P < 0.05).
The PPI network construction and hub genes selected
The STRING database (STRING, http://string-db.org) (Crosara et al. 2018) was performed to interact with the DEGs. Then, a PPI network was analyzed and visualized by the Cytoscape software (http://www.cytoscape.org/). The top 10 hub genes were selected by the plugin cytoHubba (Chin et al. 2014) (scores > 2).
Expression and prognosis analysis of hub genes in GEPIA2
GEPIA2 (http://gepia2.cancer-pku.cn/), a web-based tool, can interactively analyze the gene expression profiling of normal and cancer (Tang et al. 2019). The hub genes were reconfirmed by the boxplot analyses. The Kaplan-Meier curves assessed the prognosis of ESCC patients with hub genes.
The construction of drug-gene interaction
The QuartataWeb (http://quartata.csb.pitt.edu/) supports searching for information on drug-gene interaction (Li et al. 2020). The identified hub genes via GEPIA2 were uploaded into the database to screen existing drugs or compounds.
Statistics analysis
The moderate t-test was applied to identify DEGs. P < 0.05 was considered as statistically significant.
Results
Identification of DEGs
DEGs (2055 in GSE26886, 239 in GSE778611 and 1478 in GSE111044) were identified by GEO2R. The overlap in the three datasets contained 94 genes, as showed in the Venn diagram (Fig. 1). As a result, 13 up-regulated genes and 81 down-regulated genes were obtained (Table 2).
Gene ontology and KEGG pathway enrichment analysis
In GO terms of Biological Process (BP) annotation, it was mainly enriched in the “Positive regulation of cell proliferation”, “Skeletal system development”, “Lipoxin metabolic process”, “Proteolysis”, “Regulation of peptidase activity”, “Negative regulation of cytokine-mediated signaling pathway”, “Skin morphogenesis”, and “Keratinocyte differentiation”. In Cellular Component (CC) annotation, it was enriched in the “Extracellular exosome”, “Extracellular space”, “Extracellular region”, “Collagen type I trimer”, “Proteinaceous extracellular matrix”, and “Extracellular matrix”. In Molecular Function (MF) annotation, the “Monooxygenase activity”, “Iron ion binding”, “Serine-type peptidase activity”, “Extracellular matrix structural constituent”, “Cytokine activity”, “Platelet-derived growth factor binding”, and “Cyclin-dependent protein serine/threonine kinase regulator activity” were clustered. Additionally, DEGs did not cluster into the KEGG pathway according to the cut-off criteria (Table 3).

As shown in Fig. 2A and B, the blue ones described down-regulated genes, while the red nodes described up-regulated genes. There were 41 genes/nodes with 39 edges enriched in the network. The top 10 hub genes were: FLG, TMPRSS2, COL1A1, COL1A2, PSCA, SCEL, PPL, ACPP, CNFN, and A2ML1 (Fig. 2B). All parameters in cytoHubba were set by default.
Validation in the TCGA and GTEx projects
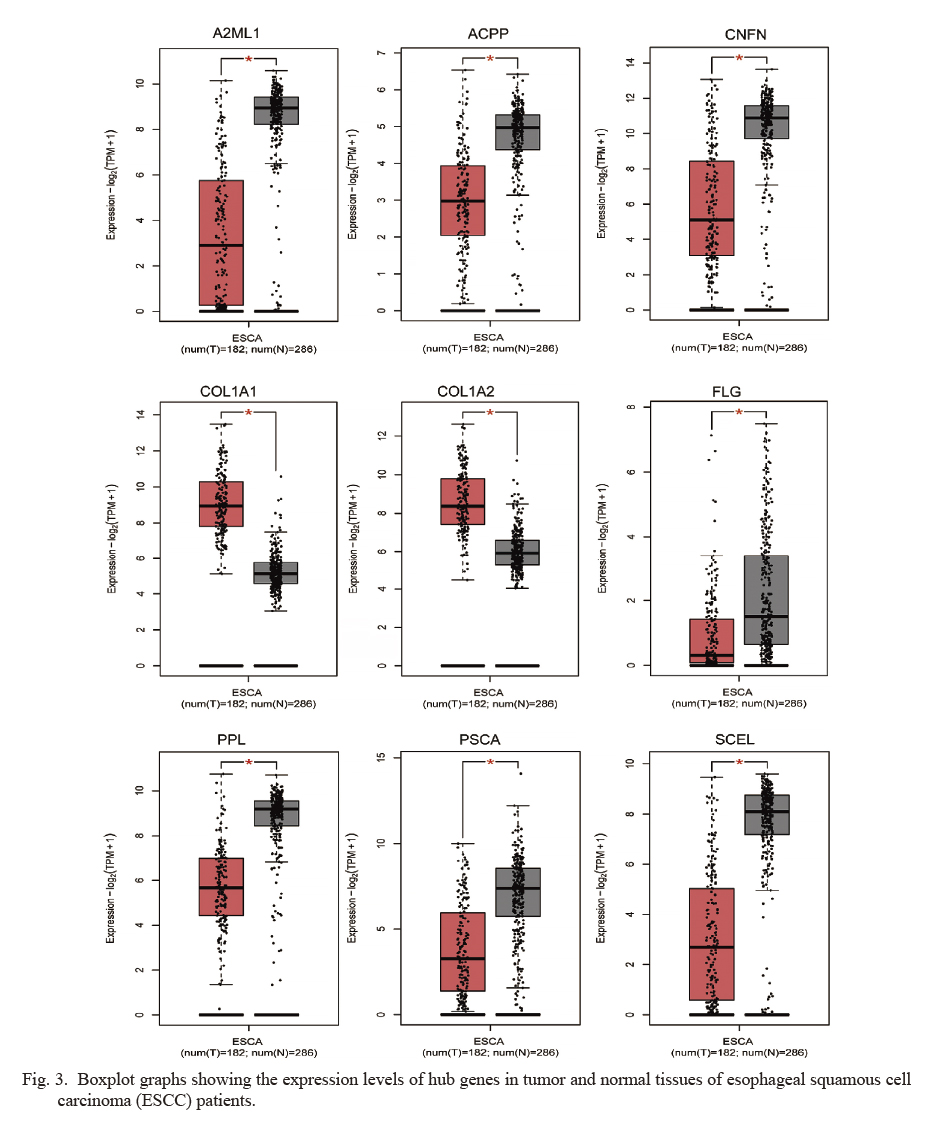
The hub genes were uploaded into the GEPIA2 website to identify the reliability of the hub genes in ESCC. Among all the ten hub genes, 9 of them (FLG, COL1A1, COL1A2, PSCA, SCEL, PPL, ACPP, CNFN, and A2ML1) were reconfirmed via GEPIA2, and the expression trends showed no change. COL1A1 and COL1A2 showed increased more significantly in ESCC tissues than in normal tissues (Fig. 3). Moreover, compared to lower COL1A1 or COL1A2 expression for ESCC patients, disease-free survival rate with higher COL1A1 or COL1A2 expression showed worse prognosis (Fig. 4), which implied that COL1A1 and COL1A2 had significant relevance to the progression of ESCC.
The construction of drug-gene interaction
The top 10 hub genes were uploaded into QuartataWeb database for drug-gene interaction analysis. As shown in Fig. 5A, just COL1A1 and COL1A2 were identified and matched to 22 predicted drugs. Among the 22 drugs, the top 5 medications approved by FDA “Collagenase clostridium histolyticum”, “Halofuginone”, “Vonicog Alfa”, “Von Willebrand Factor Human”, and “Clove oil" were selected (Table 4). Besides, COL1A1 and COL1A2 were involved in “Platelet activation”, “ECM-receptor interaction”, “PI3K-Akt signaling pathway” and others in the KEGG pathway (Fig. 5B).
Discussion
Despite the advancement of surgical and medical therapy for ESCC, the overall mortality of ESCC accounts for the sixth leading cause of cancer-related deaths (Arnold et al. 2015). High mortality in ESCC can be attributed to insufficient early-stage detection methods, chemotherapy resistance, and a high recurrence and metastasis risk (Chen et al. 2019). Therefore, the identification of reliable biomarkers for the diagnosis and treatment of ESCC is urgently required. With the rapid development of bioinformatics methods, many microarrays and sequencing data have been generated, which provide a comprehensive and convenient strategy for screening the genetic alterations and identifying the molecular mechanisms for the diagnosis and prognosis of cancers (Barrett et al. 2013; Arnold et al. 2015; Kuleshov et al. 2016; Chen et al. 2019; Li et al. 2020).
In this study, GSE26886, GSE111044 and GSE77861 were analyzed through GEO2R for identification DEGs between ESCC tissues and normal tissues. A total of 94 DEGs (13 up-regulated genes and 81 down-regulated genes) were screened. In Biological Process (BP) annotation, these genes were mainly enriched in the cell proliferation, cell migration and cell differentiation, such as “Positive regulation of cell proliferation”, “Skeletal system development”, and “Lipoxin metabolic process”. In Cellular Component (CC) annotation, these genes were mainly enriched in the “Extracellular exosome”, “Extracellular space”, “Extracellular region”, “Collagen type I trimer”, “Proteinaceous extracellular matrix”, and “Extracellular matrix”, which were closely related to the extracellular microenvironment. In Molecular Function (MF) annotation, the “Monooxygenase activity”, “Iron ion binding”, “Serine-type peptidase activity”, “Extracellular matrix structural constituent”, “Cytokine activity”, “Platelet-derived growth factor binding”, and “Cyclin-dependent protein serine/threonine kinase regulator activity” were clustered, which were related to the communication between cells and extracellular matrix. The aforementioned results suggested that the DEGs played an oncogenic role by promoting cancer cell proliferation. Additionally, DEGs did not cluster into the KEGG pathway according to the cut-off criteria.
Among the PPI construction, 41 genes/nodes with 39 edges were enriched. Additionally, the top 10 hub genes (FLG, TMPRSS2, COL1A1, COL1A2, PSCA, SCEL, PPL, ACPP, CNFN and A2ML1) were selected using cytoHubba software. Of the 10 hub genes, nine genes (FLG, COL1A1, COL1A2, PSCA, SCEL, PPL, ACPP, CNFN and A2ML1) were reconfirmed using GEPIA2, and the expression trends showed no change. COL1A1 and COL1A2 demonstrated a more significant increase in ESCC tissues than in normal tissues. Moreover, the disease-free survival rate under a higher COL1A1 or COL1A2 expression in patients with ESCC showed poor prognosis compared with that under the low COL1A1 or COL1A2 expression, which implied that COL1A1 and COL1A2 has a significant relevance to ESCC progression.
Type I collagen, which typically consists of collagen type I α 1 (COL1A1) and collagen type I α 2 (COL1A2) , is one of the components of the extracellular matrix (Yamauchi et al. 2018) and is considered to be associated with tumor invasion and progression. COL1A1 and COL1A2 exhibited an abnormal expression in some types of cancer (Zhang et al. 2018). In medulloblastoma and colorectal cancer, the COL1A1 and COL1A2 mRNA expression levels increased, whereas in melanoma and bladder cancer, the COL1A2 expression level decreased (Liu et al. 2017). A study demonstrated a correlation of high COL1A1 and COL10A1 expressions with poor prognosis in ESCC patients (Zhang et al. 2018). The disease-free survival analysis in this study indicated that ESCC patients with a high COL1A1 or COL1A2 expression showed a higher risk of disease recurrence or progression than those with a lower COL1A1 or COL1A2 expression. Meanwhile, COL1A1 and COL1A2 were enriched in “Platelet activation”, “ECM-receptor interaction” and “PI3K-Akt signaling pathway”, which play a crucial role in tumorigenesis. Therefore, COL1A1 and COL1A2 may induce the invasion and metastasis of ESCC.
Despite the vital role of COL1A1 and COL1A2 in the tumorigenesis, only a few drugs have been designed to target these two genes. In this study, we screened the old drugs to identify new anticancer drugs by using the bioinformatic approach. The ‘old drugs’ were defined as the FDA approved or those with safe clinical application. In this study, COL1A1 and COL1A2 matched to 22 predicted drugs. Among these 22 drugs, the top 5 drugs approved by FDA, namely, “Collagenase clostridium histolyticum”, “Halofuginone”, “Vonicog Alfa”, “Von Willebrand Factor Human”, and “Clove oil” were selected. “Collagenase clostridium histolyticum”, an enzyme, is produced by the bacterium Clostridium histolyticum (Brown 2017). This enzyme degrades the collagen plaques and thus is beneficial for the treatment of Dupuytren’s contracture (Grazina et al. 2019) and Peyronie’s disease (Randhawa and Shukla 2019). The topical formulation is used for the debridement of necrotic tissues due to burns or chronic ulcers. “Halofuginone” can suppress the expression of collagen a1(I) and matrix metalloproteinase 2 (MMP-2). Halofuginone can also inhibit cell proliferation and extracellular matrix deposition (Sundrud et al. 2009). “Vonicog alfa” is used for the control of bleeding episodes in patients with von Willebrand disease. It contains only the “Von Willebrand Factor Human” and thus, it offers the flexibility to administer the coagulation factor VIII if needed. The von Willebrand disease is an inherited disorder characterized by the deficiency or misfunction of the von Willebrand factor (vWF) (Singal and Kouides 2016). Due to this deficiency, the blood cannot effectively clot, and the patients having this deficiency are prone to prolonged or excessive bleeding. “Clove oil” possesses anticancer, anti-inflammatory and antioxidant properties (Cortés-Rojas et al. 2014).
Most of these drugs are used for controlling bleeding, suppressing cell proliferation and extracellular matrix deposition, and promoting blood coagulation. Meanwhile, vascular formation supplies nutrients to the tumor, which promotes development, metastasis and recurrence of the tumor (Wang et al. 2016). Probably, these drugs could show anticancer effects by promoting blood coagulation or inhibiting vascular formation in cancers, which offer a novel idea for the treatment of ESCC.
In summary, COL1A1 and COL1A2, which contribute to the tumorigenesis and prognosis of ESCC, might be a potential biomarker for ESCC diagnosis and prognosis. Additionally, the potential drugs associated with these hub genes were selected and constructed, which offer a novel idea for the treatment of ESCC. However, our interpretations are limited by considerable risk of bias and the drugs should be verified by relevant experimental models in future.
Acknowledgments
We thank all the public databases and websites used in this paper; GEO database, TCGA database, KEGG database, the Enrichr website, the Retrieval of Interacting Genes website, and the QuartataWeb.
Conflict of Interest
The authors declare no conflict of interest.
References
-
Anisimov,
V.N.
(2016) Metformin for prevention and treatment of colon cancer: a reappraisal of experimental and clinical data. Curr. Drug Targets, 17, 439-446.
-
Armstrong,
M.F.,
Lohse,
C.M.,
Lees,
K.A. &
Carlson,
M.L.
(2021) Association of metformin with volumetric tumor growth of sporadic vestibular schwannomas. Otol. Neurotol., 42, 1081-1085.
-
Arnold,
M.,
Soerjomataram,
I.,
Ferlay,
J. &
Forman,
D.
(2015) Global incidence of oesophageal cancer by histological subtype in 2012. Gut, 64, 381-387.
-
Barrett,
T.,
Wilhite,
S.E.,
Ledoux,
P.,
Evangelista,
C.,
Kim,
I.F.,
Tomashevsky,
M.,
Marshall,
K.A.,
Phillippy,
K.H.,
Sherman,
P.M.,
Holko,
M.,
Yefanov,
A.,
Lee,
H.,
Zhang,
N.,
Robertson,
C.L.,
Serova,
N.,
et al.(2013) NCBI GEO: archive for functional genomics data sets--update. Nucleic Acids Res., 41, D991-D995.
-
Brown,
R.
(2017) Recombinant von Willebrand factor for severe gastrointestinal bleeding unresponsive to other treatments in a patient with type 2A von Willebrand disease: a case report. Blood Coagul. Fibrinolysis, 28, 570-575.
-
Chen,
F.F.,
Zhang,
S.R.,
Peng,
H.,
Chen,
Y.Z. &
Cui,
X.B.
(2019) Integrative genomics analysis of hub genes and their relationship with prognosis and signaling pathways in esophageal squamous cell carcinoma. Mol. Med. Rep., 20, 3649-3660.
-
Chin,
C.H.,
Chen,
S.H.,
Wu,
H.H.,
Ho,
C.W.,
Ko,
M.T. &
Lin,
C.Y.
(2014) cytoHubba: identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol., 8 Suppl 4, S11.
-
Cortés-Rojas,
D.F.,
de Souza,
C.R.F. &
Oliveira,
W.P.
(2014) Clove (Syzygium aromaticum): a precious spice. Asian Pac. J. Trop. Biomed., 4, 90-96.
-
Crosara,
K.T.B.,
Moffa,
E.B.,
Xiao,
Y. &
Siqueira,
W.L.
(2018) Merging in-silico and in vitro salivary protein complex partners using the STRING database: a tutorial. J. Proteomics, 171, 87-94.
-
Cunha Junior,
A.D.,
Bragagnoli,
A.C.,
Costa,
F.O. &
Carvalheira,
J.B.C.
(2021) Repurposing metformin for the treatment of gastrointestinal cancer. World J. Gastroenterol., 27, 1883-1904.
-
Dalmer,
T.R.A. &
Clugston,
R.D.
(2019) Gene ontology enrichment analysis of congenital diaphragmatic hernia-associated genes. Pediatr. Res., 85, 13-19.
-
Forde,
P.M. &
Kelly,
R.J.
(2013) Chemotherapeutic and targeted strategies for locally advanced and metastatic esophageal cancer. J. Thorac. Oncol., 8, 673-684.
-
Freimanis,
G.L. &
Oade,
M.S.
(2020) Whole-genome sequencing protocols for IBV and other coronaviruses using high-throughput sequencing. Methods Mol. Biol., 2203, 67-74.
-
Grazina,
R.,
Teixeira,
S.,
Ramos,
R.,
Sousa,
H.,
Ferreira,
A. &
Lemos,
R.
(2019) Dupuytren’s disease: where do we stand? EFORT Open Rev., 4, 63-69.
-
Jang,
S.K.,
Hong,
S.E.,
Lee,
D.H.,
Kim,
J.Y.,
Kim,
J.Y.,
Hong,
J.,
Park,
I.C. &
Jin,
H.O.
(2021) Inhibition of AKT enhances the sensitivity of NSCLC cells to metformin. Anticancer Res., 41, 3481-3487.
-
Kamio,
T.,
Kamio,
H.,
Aoki,
T.,
Ondo,
Y.,
Uchiyama,
T.,
Yamamoto-Shimojima,
K.,
Watanabe,
M.,
Okamoto,
T.,
Kanno,
H. &
Yamamoto,
T.
(2020) Molecular profiles of breast cancer in a single institution. Anticancer Res., 40, 4567-4570.
-
Kanehisa,
M.,
Furumichi,
M.,
Tanabe,
M.,
Sato,
Y. &
Morishima,
K.
(2017) KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res., 45, D353-D361.
-
Kuleshov,
M.V.,
Jones,
M.R.,
Rouillard,
A.D.,
Fernandez,
N.F.,
Duan,
Q.,
Wang,
Z.,
Koplev,
S.,
Jenkins,
S.L.,
Jagodnik,
K.M.,
Lachmann,
A.,
McDermott,
M.G.,
Monteiro,
C.D.,
Gundersen,
G.W. &
Ma'ayan,
A.
(2016) Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res., 44, W90-W97.
-
Li,
H.,
Pei,
F.,
Taylor,
D.L. &
Bahar,
I.
(2020) QuartataWeb: integrated chemical-protein-pathway mapping for polypharmacology and chemogenomics. Bioinformatics (Oxford, England), 36, 3935-3937.
-
Lin,
Y.,
Totsuka,
Y.,
He,
Y.,
Kikuchi,
S.,
Qiao,
Y.,
Ueda,
J.,
Wei,
W.,
Inoue,
M. &
Tanaka,
H.
(2013) Epidemiology of esophageal cancer in Japan and China. J. Epidemiol., 23, 233-242.
-
Liu,
S.,
Liao,
G. &
Li,
G.
(2017) Regulatory effects of COL1A1 on apoptosis induced by radiation in cervical cancer cells. Cancer Cell Int., 17, 73.
-
Liu,
S.,
Lin,
Z.,
Lin,
J.,
Chen,
H.,
Xie,
Q.,
Rao,
W.,
Chen,
Y.,
Yu,
K. &
Hu,
Z.
(2021) Health-related quality of life and treatment modality among esophageal squamous cell carcinoma survivors: a prospective study using time to deterioration model analysis. J. Surg. Oncol., 123, 156-163.
-
Lordick,
F.,
Mariette,
C.,
Haustermans,
K.,
Obermannová,
R. &
Arnold,
D.
(2016) Oesophageal cancer: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann. Oncol., 27, v50-v57.
-
Marginean,
E.C. &
Dhanpat,
J.
(2020) Pathologic assessment of endoscopic resection specimens with superficial carcinoma of the esophagus: current practice and practical issues. Ann. N. Y. Acad. Sci., 1482, 130-145.
-
Morales,
D.R. &
Morris,
A.D.
(2015) Metformin in cancer treatment and prevention. Annu. Rev. Med., 66, 17-29.
-
Pillay,
S.,
Giandhari,
J.,
Tegally,
H.,
Wilkinson,
E.,
Chimukangara,
B.,
Lessells,
R.,
Moosa,
Y.,
Mattison,
S.,
Gazy,
I.,
Fish,
M.,
Singh,
L.,
Khanyile,
K.S.,
San,
J.E.,
Fonseca,
V.,
Giovanetti,
M.,
et al. (2020) Whole genome sequencing of SARS-CoV-2: adapting Illumina protocols for quick and accurate outbreak investigation during a pandemic. Genes (Basel), 11, 949.
-
Quinn,
B.J.,
Kitagawa,
H.,
Memmott,
R.M.,
Gills,
J.J. &
Dennis,
P.A.
(2013) Repositioning metformin for cancer prevention and treatment. Trends Endocrinol. Metab., 24, 469-480.
-
Randhawa,
K. &
Shukla,
C.J.
(2019) Non-invasive treatment in the management of Peyronie's disease. Ther. Adv. Urol., 11, 1756287218823671.
-
Singal,
M. &
Kouides,
P.A.
(2016) Recombinant von Willebrand factor: a first-of-its-kind product for von Willebrand disease. Drugs Today, 52, 653-664.
-
Su,
H.A.,
Hsiao,
S.W.,
Hsu,
Y.C.,
Wang,
L.Y. &
Yen,
H.H.
(2020) Superiority of NBI endoscopy to PET/CT scan in detecting esophageal cancer among head and neck cancer patients: a retrospective cohort analysis. BMC Cancer, 20, 69.
-
Sundrud,
M.S.,
Koralov,
S.B.,
Feuerer,
M.,
Calado,
D.P.,
Kozhaya,
A.E.,
Rhule-Smith,
A.,
Lefebvre,
R.E.,
Unutmaz,
D.,
Mazitschek,
R.,
Waldner,
H.,
Whitman,
M.,
Keller,
T. &
Rao,
A.
(2009) Halofuginone inhibits TH17 cell differentiation by activating the amino acid starvation response. Science, 324, 1334-1338.
-
Tang,
Z.,
Kang,
B.,
Li,
C.,
Chen,
T. &
Zhang,
Z.
(2019) GEPIA2: an enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res., 47, W556-W560.
-
Wang,
J.,
Tang,
Z.,
Wang,
S.,
Zeng,
W.,
Qian,
W.,
Wu,
L.,
Wang,
W. &
Luo,
J.
(2016) Differential diagnostic value of computed tomography perfusion combined with vascular endothelial growth factor expression in head and neck lesions. Oncol. Lett., 11, 3342-3348.
-
Xue,
J.,
Jia,
E.,
Ren,
N. &
Xin,
H.
(2021) Identification of prognostic miRNA biomarkers for esophageal cancer based on The Cancer Genome Atlas and Gene Expression Omnibus. Medicine (Baltimore), 100, e24832.
-
Yamauchi,
M.,
Taga,
Y.,
Hattori,
S.,
Shiiba,
M. &
Terajima,
M.
(2018) Analysis of collagen and elastin cross-links. Methods Cell Biol., 143, 115-132.
-
Yang,
X.,
Wang,
L.,
Du,
H.,
Lin,
B.,
Yi,
J.,
Wen,
X.,
Geng,
L. &
Du,
X.
(2021) Prognostic impact of eosinophils in peripheral blood and tumor site in patients with esophageal squamous cell carcinoma treated with concurrent chemoradiotherapy. Medicine (Baltimore), 100, e24328.
-
Ye,
H.,
Mulmi Shrestha,
S.,
Zhu,
J.,
Ding,
Y. &
Shi,
R.
(2021) Long noncoding RNA LINC00491 promotes proliferation and inhibits apoptosis in esophageal squamous cell carcinoma. Int. J. Mol. Med., 47, 1.
-
Yue,
W.,
Yang,
C.S.,
DiPaola,
R.S. &
Tan,
X.L.
(2014) Repurposing of metformin and aspirin by targeting AMPK-mTOR and inflammation for pancreatic cancer prevention and treatment. Cancer Prev. Res.. (Phila.), 7, 388-397.
-
Zhang,
X.,
Chen,
Y.,
Li,
Z.,
Han,
X. &
Liang,
Y.
(2020) TGFBR3 is an independent unfavourable prognostic marker in oesophageal squamous cell cancer and is positively correlated with Ki-67. Int. J. Exp. Pathol., 101, 223-229.
-
Zhang,
X. &
Jain,
D.
(2020) Updates in staging and pathologic evaluation of esophageal carcinoma following neoadjuvant therapy. Ann. N. Y. Acad. Sci., 1482, 163-176.
-
Zhang,
Z.,
Wang,
Y.,
Zhang,
J.,
Zhong,
J. &
Yang,
R.
(2018) COL1A1 promotes metastasis in colorectal cancer by regulating the WNT/PCP pathway. Mol. Med. Report., 17, 5037-5042.