Abstract
Although splenomegaly is one of the important signs of primary myelofibrosis, the differential diagnosis varies from malignant disorders to benign disorders, including malignant lymphoma and sarcoidosis. The patient was a 67-year-old male who developed anemia and huge splenomegaly. The laboratory findings include human T-cell leukemia virus type 1 (HTLV-1) antibody, elevated soluble interleukin-2 receptor, hypocellular bone marrow, and uptake in the spleen on positron emission tomography/computed tomography scan. Additionally, we performed laparoscopic splenectomy to alleviate the clinical symptoms and to rule out malignant lymphoma. Histological findings revealed extramedullary hematopoiesis, characterized by the presence of erythroid islands and clusters of dysplastic megakaryocytes with increased reticulin fibrosis. Immunohistochemical staining revealed the presence of von Willebrand factor, dysplastic megakaryocytes, myeloperoxidase, myeloid-predominant proliferations, and CD34 immature myeloid cells. Furthermore, regarding the angiogenesis in the spleen, the endothelial cells of the capillaries and those of the sinusoidal vascular system that were reactive for CD34 and CD8, respectively, were also detected. Consequently, the histological findings revealed both extramedullary hematopoiesis and angiogenesis in spleen. Based on the histological findings and the identification of Janus activating kinase 2 (JAK-2) mutation, the patient was diagnosed with primary myelofibrosis. Splenectomy reduces blood transfusion requirements after surgery. The patient was carefully followed-up without further treatments. Thus, primary myelofibrosis is the crucial differential diagnosis of huge splenomegaly.
Introduction
Primary myelofibrosis (PMF) is a chronic progressive stem cell disorder of myeloproliferative disorders (MPDs) characterized by anemia, thrombocytopenia, splenomegaly, and fibrosis in the bone marrow (BM) (Barbui et al. 2018; Garmezy et al. 2021). For the past decades, significant progress has been achieved in the field of MPDs (Barbui et al. 2018; Garmezy et al. 2021). Notably, driver mutations in myeloproliferative neoplasms (MPNs), including Janus activating kinase 2 (JAK-2), calreticulin (CALR), and thrombopoietin receptor (MPL) gene mutations, play a major role in the pathogenesis of MPDs (Barbui et al. 2018; Garmezy et al. 2021). However, no specific pathological gene mutations have been identified in PMF (Barbui et al. 2018; Garmezy et al. 2021). To date, PMF has been clinically diagnosed according to the World Health Organization criteria, including pathological findings, exclusion of other MPDs, genetic mutations, and clinical findings (Barbui et al. 2018; Garmezy et al. 2021). Although splenomegaly is a significant finding of PMF, the differential diagnosis varied from malignant disorders to benign disorders, including malignant lymphoma and sarcoidosis (Shimoda et al. 2020). Furthermore, PMF belongs to a group of diseases known as the MPNs that makes it difficult to make precise clinical and pathological diagnosis due to the rare incidence and diverse clinical manifestations at the stage of myelofibrosis (Barbui et al. 2018; Garmezy et al. 2021). In Japan, an epidemiologic prospective analysis of PMF during 17 years, from 1999 to 2015, showed 780 patients only (Shimoda et al. 2020); thus, the clinical features and outcomes are unclear due to the limited previous reports. In particular, Klco et al. (2010) reported that the diagnosis of PMF may be problematic at diverse clinical manifestations according to the stage of myelofibrosis. In the early stage, most PMF patients have a nonspecific, usually multilineage, expansion of hematopoiesis with minimal fibrosis and neutrophilic leukocytosis and thrombocytosis (Klco et al. 2010). However, as myelofibrosis increases, the peripheral blood count frequently declines, and patients become symptomatic with fatigue, bleeding, and infections due to cytopenia (Klco et al. 2010). Therefore, splenomegaly is marked as a result of extramedullary hematopoiesis (Klco et al. 2010).
In this study, we describe a rare case of primary myelofibrosis diagnosed with huge splenomegaly and treated with laparoscopic splenectomy. The histological findings after laparoscopic splenectomy assist in the diagnosis of MPDs.
Case Presentation
The patient was a 67-year-old male who developed general fatigue in January 2015. The patient was initially diagnosed with anemia and splenomegaly and was referred to our hospital for further examination. Upon admission, the patient showed huge splenomegaly with no systemic lymphadenopathy. Laboratory findings are shown in Table 1. Laboratory findings showed anemia (hemoglobin 6.9 g/dl) with normal leukocytes (white blood cells 5.640 × 103/μL), normal platelet counts (19.4 × 104/μL), positive human T-cell leukemia virus type 1 (HTLV-1) antibody and slight elevation of soluble interleukin-2 receptor (sIL-2R; 786 IU/L). Abdominal computed tomography (CT) revealed huge splenomegaly without any systemic lymph node swelling (Fig. 1A). Positron emission tomography (PET)/CT scan showed slight uptake in the spleen (SUV 3.9) (Fig. 1B). BM aspiration showed hypocellular marrow with slight dysplasia in the erythroid series (Fig. 1C). Flow cytometry (FCM) analysis revealed the absence of an abnormal phenotype of blasts, and chromosomal analysis revealed a normal karyotype (46, XY). Furthermore, a chimeric screening test for leukemia revealed negative findings.
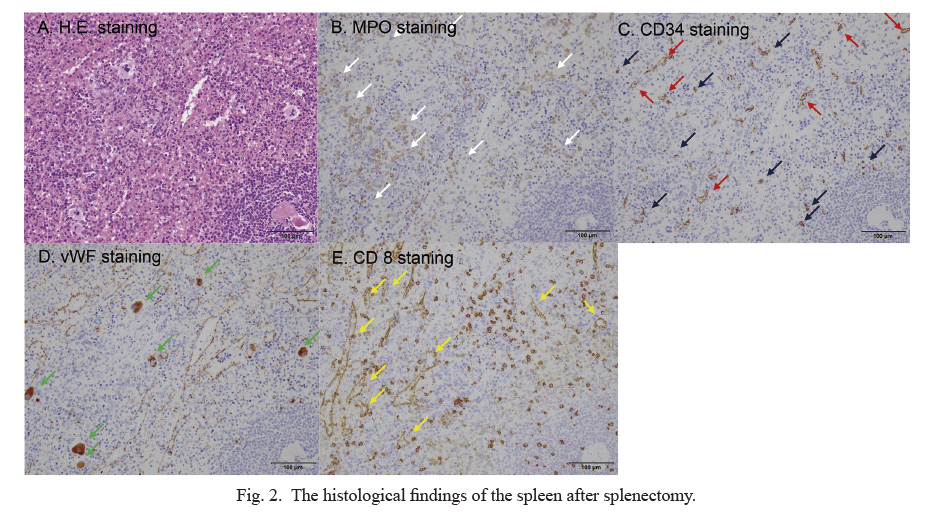
The patient showed huge splenomegaly, complicating the dilatation of the splenic artery and vein and compression of the kidney and abdominal artery. To alleviate the clinical symptoms, including abdominal compression, and to rule out malignant lymphoma, we performed laparoscopic splenectomy as the therapeutic diagnosis. The histological findings revealed extramedullary hematopoiesis in the spleen, suggesting the presence of MPDs (Fig. 2A). In addition, immunohistochemical staining revealed the presence of myeloperoxidase (MPO) positive myeloid predominant proliferations (Fig. 2B, white arrows), CD34 positive immature myeloid cells (Fig. 2C, blue arrows) and von Willebrand factor positive dysplastic megakaryocytes (Fig. 2D, green arrows). The patient had an anti-HTLV-1 antibody. Furthermore, we also investigated the clonality of HTLV-1 in the spleen after splenectomy. Southern blot analysis showed no clonal band of adult T-cell leukemia-lymphoma (ATL) cells, suggesting HTLV-1 carrier status in the patient. Therefore, histopathological findings play a major role in assisting the diagnosis of MPD.
In addition, we performed a BM biopsy. The histological findings showed myelofibrosis by Hematoxylin and Eosin staining, Silver staining, and Azan staining (Fig. 3A-C; black arrows). Furthermore, CD41 positive megakaryocytes with dysplasia. (Fig. 3D; green arrows) and CD34 positive immature myeloid cells (Fig. 3E; blue arrows) were detected. Thus, the findings of BM were classified as MF-3 by the European Consensus on Grading Bone Marrow Fibrosis (Shimoda et al. 2020). Janus activating kinase 2 (JAK-2) mutation was identified by genetic analysis. Taken these findings together, the patient was diagnosed with PMF.
We retrospectively examined the peripheral blood smear and BM smears of the patient. The peripheral blood smear revealed the presence of small amount of dacryocytes and polychromatophilic erythrocytes (Fig. 1D). Moreover, our study suggested that a BM biopsy should have been performed before splenectomy when a hematologic disease was suspected in patients with isolated splenomegaly.
Consequently, splenectomy reduces blood transfusion requirements after surgery without further medication. According to the Dynamic International Prognostic Scoring System (DIPSS) (Shimoda et al. 2020), the patient in this study was scored as 3. If the score is 2-3, the patient is considered as “Intermediate-2 risk.” Median survival was 4 years with 50% survival from the time of diagnosis. However, there is no consensus on the indication of JAK-2 inhibitors for the treatment of PMF after splenectomy (Santos et al. 2014; Kitanaka et al. 2016; Shimoda et al. 2020). Therefore, without medication, the patient was carefully monitored.
Angiogenesis is the generation of new blood vessels from the pre-existing vasculature. In PMF, the angiogenesis in the spleen generates the endothelial cells of capillaries and endothelial cells of the sinusoidal vascular system. In this study, immunochemical staining was used to differentiate the new blood vessels. The endothelial cells of capillaries and those of the sinusoidal vascular system that were reactive for CD34 (Fig. 2C; red arrows) and CD8 (Fig. 2E; yellow arrows), respectively, were detected. Therefore, these findings are consistent with the presence of angiogenesis in the spleen. Thus, the histological findings revealed both the extramedullary hematopoiesis and the angiogenesis in the spleen of PMF.
The study was approved by an institutional ethics committee (19-12). The study was conducted in accordance with the Declaration of Helsinki 1975. A statement regarding this effect has been included in the manuscript.
Discussion
In this case report, we described a rare case of PMF with diagnosed splenomegaly and treated with laparoscopic splenectomy. This study corresponded to the fibrotic stage of PMF, which presented with anemia and splenomegaly without leukocytosis and thrombocytosis. Furthermore, histological findings of the spleen showed both the presence of the extramedullary hematopoiesis and the angiogenesis in the spleen. Therefore, pathological findings of PMF in patients show some merits for clinical hematologists.
In contrast to previous reports (Mesa et al. 2001; Barosi et al. 2004), we recently focused on splenic extramedullary hematopoiesis. Splenic extramedullary hematopoiesis plays a major role in the pathogenesis and outcome of PMF.
Extramedullary hematopoiesis is considered to be a consequence of sequestration, accumulation, and proliferation of circulating progenitor cells in extramedullary sites (Mesa et al. 2001; Barosi et al. 2004). However, the histological findings and pathogenesis of PMF remain unclear. In this study, the histological findings were presented with extramedullary hematopoiesis and support the presence of MPDs. Barosi et al. (2004) reported that splenomegaly in MPN including MF is acknowledged as the result of splenic extramedullary hematopoiesis. Furthermore, as documented by the measurement of capillary vascular density, neoangiogenesis significantly contributes to the enlargement of the spleen, (Barosi et al. 2004). Consistent with the previous findings (Barosi et al. 2004), immunohistochemical analysis revealed the presence of von Willebrand factor, dysplastic megakaryocytes, myeloperoxidase (MPO), myeloid predominant proliferations, and CD34 immature myeloid cells.
Furthermore, our study revealed the detection of the endothelial cells of the capillaries and those of the sinusoidal vascular system that are reactive for CD8 and CD34, respectively, suggesting the angiogenesis in PMF. Consequently, histopathological findings may assist in the diagnosis of MPD. Next, we compared the differences of histological findings among seven cases with huge splenomegaly that we encountered in our institution (Table 2). Among the seven cases, five (case 1, 2, 3, 4, and 6) showed both the proliferation of lymphoma cells and the presence of angiogenesis. One case (case 5) showed a non-caseating epithelioid cell granuloma with the presence of angiogenesis. The present case also showed both the extramedullary hematopoiesis and the angiogenesis in spleen. Thus, the angiogenesis may be essential to support the proliferation of lymphoma cells, non-caseating epithelioid cell granuloma and extramedullary hematopoiesis in the spleen. However, in our case, we did not perform a quantitative analysis of the extent to which CD34+ capillaries and CD8+ sinus endothelial cells occupy extramedullary hematopoiesis. Furthermore, we did not evaluate the proliferation ability of both endothelial cells using the labeling index of Ki-67. Therefore, it is unclear how much angiogenesis plays a significant role in splenomegaly. In the future, further studies are essential to reveal the relationship between angiogenesis and splenomegaly in MPDs.
Recent studies have revealed that splenomegaly affects the overall survival (OS) of PMF (Vannucchi et al. 2015; Song et al. 2016). Vannucchi et al. (2015) reported that a larger spleen size at baseline was prognostic for shortened survival, and reductions in spleen size with ruxolitinib treatment correlated with longer survival. Furthermore, Song et al. (2016) reported the usefulness of spleen volume measured by computed tomography for predicting clinical outcomes in 188 PMF patients. Furthermore, Song et al. (2016) revealed that PMF patients with low spleen volume status had superior leukemia-free survival and overall survival compared to those with high spleen volume status. According to the DiPSS, in this study, the patient was scored as 3. If scored 2-3, the patient is considered as “intermediate risk-2”. Median survival was four years, with 50% survival from the time of diagnosis. Therefore, in this study, the OS might be poorer than that of the usual intermediate risk-2.
Furthermore, regarding huge splenomegaly patients, Shimizu-Kohno et al. (2012) retrospectively analyzed 193 cases of spleen specimens after splenectomy. Among 193 cases, lymphoid neoplasm consisted of 64.2% (n = 124), other neoplasms consisted of 9.4% (n = 18), and non-neoplasms comprised 26.4 % (n = 51). Among these 193 cases, MPDs accounted for only 1.6% (n = 3). Therefore, this retrospective study revealed the rare incidence of MPDs in patients with huge splenomegaly (Shimizu-Kohno et al. 2012). In our institution, we encountered seven cases with huge splenomegaly (Table 2). All cases showed isolated splenomegaly without systemic lymph node swelling (Table 2). Furthermore, six out of seven patients showed no mass-forming isolated splenomegaly on CT scan (Table 2). To alleviate the clinical symptoms, including abdominal compression, we performed laparoscopic splenectomy as the therapeutic diagnosis. Among the eight cases, six were diagnosed with malignant lymphoma. The other two cases were diagnosed with splenic sarcoidosis (Kawano et al. 2012) and PMF in the present case, respectively. Therefore, histopathological findings assist in the diagnosis of patients with splenomegaly.
The position of splenectomy for PMF treatment has decreased due to the development of new agents, like JAK-2 inhibitors (Shimoda et al. 2020). Moreover, our study suggested that a BM biopsy should have been performed before splenectomy when a hematologic disease was suspected in patients with isolated splenomegaly. However, in this study, lymphoma and sarcoidosis were unlikely to be the differential diagnoses due to relatively low serum sIL-2R levels and the lack of specific findings on chest X-ray. Furthermore, it is advisable to convey our experiences from this study to guide in the diagnosis of underlying diseases like isolated splenomegaly.
To date, the mechanisms of PMF, including the characteristics and behaviors of CD34+ malignant stem cells, the roles of cytokines, and the origins of fibroblast cells have been illustrated in an in vivo mouse model ( Wang et al. 2012; Ozono et al. 2021; Saito et al. 2021). Wang et al. (2012) reported the successful engraftment of human splenic CD34+ cells as donor cells in a recipient mouse, showing that the spleens of myelofibrosis patients contain malignant hematopoietic stem cells. In Jak2V617F-induced primary myelofibrosis mice, Ozono et al. (2021) revealed that neoplastic fibrocytes are the major contributors to BM fibrosis in PMF, and TGF-β1 is required for their differentiation. Furthermore, in a xenograft mouse model using primary PMF specimens, Saito et al. (2021) reported that CD34+ malignant PMF stem cells directly differentiate into fibroblast-like cells and cytokine factors, including TGF that affect the mesenchymal lineage cells into fibrotic changes in the BM. Therefore, the elucidation of PMF pathogenesis may assist in the development of new therapeutic agents.
In conclusion, this study aims to emphasize the significance of PMF for the differential diagnosis of patients with huge splenomegaly. Clinical findings of the patient should preferably be taken into consideration before performing laparoscopic splenectomy.
Acknowledgments
We thank the medical staff, including nurses, laboratory technicians, pharmacists, physical therapists, mental therapists, psychiatrists, and nutritionists for their excellent care of the patient.
Conflict of Interest
The authors declare no conflict of interest.
References
-
Barbui,
T.,
Thiele,
J.,
Gisslinger,
H.,
Kvasnicka,
H.M.,
Vannucchi,
A.M.,
Guglielmelli,
P.,
Orazi,
A. &
Tefferi,
A.
(2018) The 2016 WHO classification and diagnostic criteria for myeloproliferative neoplasms: document summary and in-depth discussion. Blood Cancer J., 8, 15.
-
Barosi,
G.,
Rosti,
V.,
Massa,
M.,
Viarengo,
G.L.,
Pecci,
A.,
Necchi,
V.,
Ramaioli,
I.,
Campanelli,
R.,
Marchetti,
M.,
Bazzan,
M. &
Magrini,
U.
(2004) Spleen neoangiogenesis in patients with myelofibrosis with myeloid metaplasia. Br. J. Haematol., 124, 618-625.
-
Garmezy,
B.,
Schaefer,
J.K.,
Mercer,
J. &
Talpaz,
M.
(2021) A provider’s guide to primary myelofibrosis: pathophysiology, diagnosis, and management. Blood Rev., 45, 100691.
-
Kawano,
S.,
Kato,
J.,
Kawano,
N.,
Yoshimura,
Y.,
Masuyama,
H.,
Fukunaga,
T.,
Shimao,
Y.,
Mihara,
K.,
Ueda,
A.,
Toyoda,
K.,
Imamura,
T. &
Kitamura,
K.
(2012) Sarcoidosis manifesting as cardiac sarcoidosis and massive splenomegaly. Intern. Med., 51, 65-69.
-
Kitanaka,
A.,
Takenaka,
K.,
Shide,
K.,
Miyamoto,
T.,
Kondo,
T.,
Ozawa,
K.,
Kurokawa,
M.,
Akashi,
K. &
Shimoda,
K.
(2016) Splenic irradiation provides transient palliation for symptomatic splenomegaly associated with primary myelofibrosis: a report on 14 patients. Int. J. Hematol., 103, 423-428.
-
Klco,
J.M.,
Vij,
R.,
Kreisel,
F.H.,
Hassan,
A. &
Frater,
J.L.
(2010) Molecular pathology of myeloproliferative neoplasms. Am. J. Clin. Pathol., 133, 602-615.
-
Mesa,
R.A.,
Li,
C.Y.,
Schroeder,
G. &
Tefferi,
A.
(2001) Clinical correlates of splenic histopathology and splenic karyotype in myelofibrosis with myeloid metaplasia. Blood, 97, 3665-3667.
-
Ozono,
Y.,
Shide,
K.,
Kameda,
T.,
Kamiunten,
A.,
Tahira,
Y.,
Sekine,
M.,
Akizuki,
K.,
Nakamura,
K.,
Iwakiri,
H.,
Sueta,
M.,
Hidaka,
T.,
Kubuki,
Y.,
Yamamoto,
S.,
Hasuike,
S.,
Sawaguchi,
A.,
et al.(2021) Neoplastic fibrocytes play an essential role in bone marrow fibrosis in Jak2V617F-induced primary myelofibrosis mice. Leukemia, 35, 454-467.
-
Saito,
N.,
Yamauchi,
T.,
Kawano,
N.,
Ono,
R.,
Yoshida,
S.,
Miyamoto,
T.,
Kamimura,
T.,
Shultz,
L.D.,
Saito,
Y.,
Takenaka,
K.,
Shimoda,
K.,
Harada,
M.,
Akashi,
K. &
Ishikawa,
F.
(2021). Circulating CD34+CD38- cells of primary myelofibrosis patients contribute to myeloid dominant hematopoiesis and bone marrow fibrosis in immunodeficient mice. Int. J. Hematol., doi: 10.1007/s12185-021-03239-y. [Epub ahead of print].
-
Santos,
F.P.,
Tam,
C.S.,
Kantarjian,
H.,
Cortes,
J.,
Thomas,
D.,
Pollock,
R. &
Verstovsek,
S.
(2014) Splenectomy in patients with myeloproliferative neoplasms: efficacy, complications and impact on survival and transformation. Leuk. Lymphoma, 55, 121-127.
-
Shimizu-Kohno,
K.,
Kimura,
Y.,
Kiyasu,
J.,
Miyoshi,
H.,
Yoshida,
M.,
Ichikawa,
R.,
Niino,
D. &
Ohshima,
K.
(2012) Malignant lymphoma of the spleen in Japan: a clinicopathological analysis of 115 cases. Pathol. Int., 62, 577-582.
-
Shimoda,
K.,
Takahashi,
N.,
Kirito,
K.,
Iriyama,
N.,
Kawaguchi,
T. &
Kizaki,
M.
(2020) JSH practical guidelines for hematological malignancies, 2018: I. Leukemia-4. Chronic myeloid leukemia (CML)/myeloproliferative neoplasms (MPN). Int. J. Hematol., 112, 268-291.
-
Song,
M.K.,
Chung,
J.S.,
Lim,
S.N.,
Lee,
G.W.,
Lee,
S.M.,
Lee,
N.K.,
Choi,
J.C. &
Oh,
S.Y.
(2016) Usefulness of spleen volume measured by computed tomography for predicting clinical outcome in primary myelofibrosis. Int. J. Hematol., 104, 476-484.
-
Vannucchi,
A.M.,
Kantarjian,
H.M.,
Kiladjian,
J.J.,
Gotlib,
J.,
Cervantes,
F.,
Mesa,
R.A.,
Sarlis,
N.J.,
Peng,
W.,
Sandor,
V.,
Gopalakrishna,
P.,
Hmissi,
A.,
Stalbovskaya,
V.,
Gupta,
V.,
Harrison,
C.,
Verstovsek,
S.,
et al.(2015) A pooled analysis of overall survival in COMFORT-I and COMFORT-II, 2 randomized phase III trials of ruxolitinib for the treatment of myelofibrosis. Haematologica, 100, 1139-1145.
-
Wang,
X.,
Prakash,
S.,
Lu,
M.,
Tripodi,
J.,
Ye,
F.,
Najfeld,
V.,
Li,
Y.,
Schwartz,
M.,
Weinberg,
R.,
Roda,
P.,
Orazi,
A. &
Hoffman,
R.
(2012) Spleens of myelofibrosis patients contain malignant hematopoietic stem cells. J. Clin. Invest., 122, 3888-3899.