Abstract
Since the middle of the last century, there have been amazing therapeutic advances for hemophilia such as the development of plasma-derived products and bioengineered recombinant factors VIII and IX (for hemophilia A and B, respectively) with improved stability, higher activity, and extended half-life. The recent use of a monoclonal antibody that mimics factor VIII activity (which is an efficient treatment for all hemophilia A phenotypes with or without inhibitors) has shown the great possibilities of non-factor therapies for improving the quality of life of hemophilia A patients, with a safer application and long-lasting effects. Gene therapy offers the promise of a “true cure” for hemophilia based on the permanent effect that a gene edition may render. Clinical trials developed in the last decade based on adenoviral vectors show modest but consistent results; now, CRISPR/Cas technology (which is considered the most efficient tool for gene edition) is being developed on different hemophilia models. Once the off-target risks are solved and an efficient switch on/off for Cas activity is developed, this strategy might become the most feasible option for gene therapy in hemophilia and other monogenic diseases.
Introduction
Hemophilia is characterized by a functional deficiency of factor VIII (FVIII) or factor IX (FIX) secondary to pathogenic variants in the F8 (Hemophilia A, HA) or F9 (Hemophilia B, HB) gene whose respective loci are located near the Xq telomere region. Both diseases are X-linked recessive traits and typically affect males while carrier females are non-symptomatic. According to the plasma concentrations of functional proteins, the disorder is classified as mild (> 5-40 IU/dL), moderate (1-5 IU/dL), or severe (< 1 IU/dL) (Blanchette et al. 2014).
Worldwide, 324,648 patients with a bleeding disorder (hemophilia, von Willebrand disease, or other rare diseases) were recently reported by the World Federation of Hemophilia. Of this total, 195,263 persons were diagnosed with hemophilia (World Federation of Hemophilia 2019). Nonetheless, a meta-analysis based on the national registries from six high-income countries, establishes a higher prevalence of hemophilia than that previously estimated. If we consider the global population of 7.5 billion inhabitants (3.8 billion males), a prevalence at birth (per 100,000 males) of 17 cases for all severities of and four cases of HB, and the inherent life expectancy disadvantage (life lost, years of life with disability and disease burden), almost 794,000 males could be hemophiliacs, including about 270,000 severely affected (Iorio et al. 2019; World Federation of Hemophilia 2019).
Without appropriate treatment, life expectancy of severely ill patients is reduced by 10 years compared with the general population. Prophylaxis based on protein substitution therapy (PST), through intravenous administration of recombinant or plasma-derived clotting factors, is considered a gold standard to avoid spontaneous bleeding episodes (Evens et al. 2018). Absence of PST triggers repetitive bleeding and chronic injuries and results in a long recovery period that affects the daily activities of patients (Guo et al. 2019). PST is the current treatment for hemophilia and has important advantages like easy administration, prolonged coagulant activity, and safety. However, PST has the following drawbacks: a short half-life (12-24 hours) requiring frequent dose administration; development of inhibitors against plasma-derived or recombinant proteins; a very high cost (approximately $300,000 US per year in adults) (High et al. 2014); and more importantly, PST is not a cure for hemophilia (Evens et al. 2018).
This narrative review provides a general outlook of the amazing therapy development for hemophilia with an emphasis on gene therapy approaches. In the last two decades, novel therapies have been developed by bioengineering to provide stable and safe expression of the deficient FVIII and FIX proteins. Alternative approaches to PST have emerged; for instance, monoclonal antibodies that mimic the FVIII function, gene therapy through viral vectors or DNA plasmids, and gene edition with enzyme systems are some strategies aimed to “cure” hemophilia. However, in the case of adeno-associated virus (AAV), which are the most popular viral vectors for gene therapy in hemophilia, preexisting viral infections with some serotypes may prevent a considerable number of individuals from the general population from receiving this strategy. Additionally, known and unknown immune responses, cellular stress, and possible random integration of viral vectors continue to challenge the provision of safe gene therapy (Weyand and Pipe 2019). New experimental models for gene edition that use the clustered regularly interspersed palindromic repeats (CRISPR) and their associated Cas proteins (CRISPR/Cas system), as alternative strategy to viral vectors, promise to provide an effective cure for hemophilia patients in the years to come (Fig. 1).

Current Management and Treatments in Hemophilia
The prophylactic infusion of factor concentrates is the most widely employed treatment for severely ill pediatric and adult patients with hemophilia (PWH). For HA and HB, the specific concentrates aim to achieve hemostatic levels of circulating FVIII or FIX to reduce or even avoid spontaneous bleeding events. An increase of at least 1% of circulating clotting factor activity in such patients is essential to prevent bleeding episodes; however, aspects like product type (recombinant or plasma-derived factors), pharmacokinetic (PK) parameters (frequency and magnitude of activity peaks), and the patient’s biology may influence hemostatic efficiency and ultimately determine a successful treatment (Hermans and Dolan 2020).
The recently developed “next-generation proteins” seek to extend the half-life (EHL) of therapeutic coagulant factors. These drugs have the potential to remain active for a longer duration in plasma; therefore, the factor infusion frequency decreases considerably. An alternative approach to EHL proteins stimulates hemostasis via non-factor therapies; for instance, monoclonal antibodies are a valuable choice in patients with or without inhibitors because a dose administered subcutaneously once per month avoids bleeding episodes and considerably enhances the patient’s life quality (Morfini and Marchesini 2020).
Next-Generation Recombinant Factors
In the early 2010s, EHL factors were engineered from recombinant proteins that underwent two main modifications: A) the fusion of fragments from other proteins like Fc of immunoglobulin G (IgG) or albumin; and B) the addition of polymers like polyethylene glycol (PEG) (Mannucci 2020). Neonatal Fc receptor enables factor recycling in plasma and prolongs the recirculation and effective activity of EHL factors (Dumont et al. 2012; Schulte 2013) while the slow degradation and renal elimination of PEG further enhance the maintenance and recirculation of such complexes (Ivens et al. 2013; Swierczewska et al. 2015). While standard recombinant factors are generally administrated twice per week, EHL concentrates can be given once per week or less depending on the frequency of bleeding episodes (Collins et al. 2016).
Fc-fusion domains
The Fc receptor binding ability is expressed in the endothelial cells of the vasculature and offers protection from endocytosis and lysosomal degradation. Recombinant FVIII Fc-fusion protein (rFVIIIFc) that joins a FVIII molecule with a Fc domain of human IgG1 was the first EHL protein approved as a prophylaxis treatment for HA patients in the European Union and the United States (Powell et al. 2012; Mahlangu et al. 2014). The rFVIIIFc circulation/long-term efficacy and safety have been documented in several clinical trials (Morfini and Marchesini 2020).
Albumin-fusion molecules
Albumin is the most abundant protein in plasma and has been used as a ligand in EHL factors for hemophilia, with an average of 20 days of activity (Santagostino et al. 2012). Recombinant fusion of FVIII and FIX factors with albumin is designed to improve the coagulation factor activity for HA or HB patients with inhibitors. The molecule complex is produced by the fusion of the wild-type factor and recombinant albumin through a linker produced in Chinese hamster ovary cells. The modification of the wild amino acid sequence of the coagulation factors is not required to produce the fusion protein; besides, the protein complex can simulate the wild-type FVIII or FIX protein’s activity (Negrier 2016; Escobar et al. 2019).
PEGylation
PEGylation or addition of PEG molecules confers a slow degradation of coagulant proteins in plasma. PEGylation addition can be site-specific or random (Mancuso and Santagostino 2017) and improves the half-life in comparison to FVIII and FIX recombinant factors. While PEGylation has demonstrated a better PK profile, some cellular effects are associated with long-term PEG exposition; for instance, PEG vacuole formation in choroid plexus cells of the blood-brain barrier but without gross cellular damage (Escobar et al. 2019).
PEGylation, Fc-fusion domains, and albumin-fusion molecules are on the way to replace the conventional PST due to their hemostatic regulation ability and extended half-life in plasma (Croteau et al. 2021). However, some next-generation factor assays (albumin-fusion or PEGylation) report that those molecules may interfere with the normal extravascular distribution of coagulation factors or the development of antibodies against the complementary molecule epitopes and hence generate clinical concerns due to the discordance between bleeding symptoms and factor activity in patients with HB (Kleiboer et al. 2020; Malec et al. 2020). Therefore, further studies are required to test their safety as a prophylactic treatment.
Emicizumab
HA patients have another option beyond traditional PST with non-factor therapies. Designed to replace the activated FVIII (FVIIIa) function, Emicizumab (HEMLIBRA®, Roche, Genentech, Inc., South San Francisco, CA, USA) is a humanized bispecific monoclonal antibody that supports the spatial interaction between activated FIX (FIXa) and FX and promotes thrombin formation by mimicking FVIIIa activity. It has demonstrated excellent efficacy with limited adverse effects in HA patients with and without inhibitors (HAVEN clinical trials) (Oldenburg et al. 2017; Mahlangu et al. 2018; Young et al. 2019) and is currently used in all HA cases regardless of their FVIII level, inhibitor presence, age, or bleeding severity (Kitazawa et al. 2012; Manucci 2020).
Prophylactic subcutaneous administration of Emicizumab (HEMLIBRA®) has demonstrated clinical efficacy despite the inability of coagulation assays to monitor or quantify its hemostatic effect. Emicizumab has been well tolerated by patients, but its use in combination with other bypass agents like activated prothrombin complex concentrates (aPCCs) is not recommended due to increased thrombotic risk (Hartmann et al. 2018). The main characteristics of all current modalities for hemophilia treatment are described in Table 1.
Despite all advantages of next-generation factors, it is necessary to develop a permanent cure for hemophilia patients (VandenDriessche and Chuah 2017). The potential of gene therapy to correct or modify pathogenic variants in vitro through vector or enzyme strategies is expected to provide effective and long-lasting treatments for hemophilia and other monogenic diseases. Hemophilia, as a single-gene disease, is an excellent candidate for gene therapy (Guo et al. 2019) aimed to deliver a long-life treatment via a unique intervention (Mannucci 2020) (Fig. 1). Although EHL factors have demonstrated an elongated pharmacokinetic propriety, compared to standard recombinant factors, they have still to be tested in a clinical study (Preijers et al. 2021).
Hemophilia-Like Model Disease for Gene Therapy
The main purpose of gene therapy is to correct diseases caused by gene dysfunctions. This technology is applied to several human diseases like cancer and cardiovascular and neurodegenerative disorders; however, the most promising application is on monogenic diseases associated with a well-characterized defective gene like hemophilia. Under this principle, the integration of a normal coding sequence into the genome of patients with severe hemophilia (ex vivo therapy) could result into a moderate or mild phenotype (Guo et al. 2019). Hence, hemophilia is the perfect candidate for gene therapy. The goal of this single-step strategy is to obtain a stable high-level expression of circulating coagulation factors (FVIII and FIX) and thus correct the hemorrhagic phenotype throughout life (Mannucci 2020).
Not only does a “true cure” for hemophilia require the introduction of a coding sequence but it is also necessary to select an appropriate delivery strategy depending on the target cells to enable the cassette expression for FVIII or FIX production. Also, the therapeutic gene must be integrated into a specific locus and the target cells should be non-dividing post-mitotic cells (e.g., hepatocytes or skeletal muscle cells). Additionally, the need for immune tolerance induction to coagulation factors after gene therapy depends on several variables like vector design, target cells, and pathogenic variant (Evens et al. 2018). Alternatively, the F8 or F9 gene could be delivered through proper vectors into stem/progenitor cells with convenient differentiation-proliferative capacity and immunoregulatory proprieties (Olmedillas López et al. 2016).
AAV Vectors on Hemophilia Treatment
Adeno-associated virus (AAV) is the most used viral vector for gene therapy in hemophilia. AAV is a non-enveloped parvovirus capable of safely delivering DNA into cells and generating recombinant molecules with eukaryotic genes that will produce the corresponding proteins. Because these viruses have a limited packing capacity of up to five kb of DNA and their integration efficiency into the host-cell genome is restricted only to the AAVS1 locus on chromosome 19 (Kotin et al. 1992), the resulting gene expression is often transitory especially on active dividing cells (Asokan et al. 2012).
The vectors AAV 2/8 and AAV5 with several modifications to improve the specificity and infection of target cells are usually used in hemophilia (Croteau et al. 2021). The first AAV clinical trial involved ten patients with severe HB at the Royal Free Hospital, London, UK. Six of them received a single high dose of AAV8 with a current follow-up of four years and have shown a stable transgene expression with FIX plasma levels between 2% and 5% as well as reduced bleeding episodes (Nathwani et al. 2014; Mannucci 2020). Among modified AAV vectors, the FIX-Padua variant (FIXR338L) confers FIX coagulant hyperactivity (approximately 8-fold) as compared with wild-type FIX (Monahan 2015). Indeed, gene therapy with the Padua variant (FIXR388L) has shown stable and prolonged protein expression (33.7%) in plasma for at least 52 weeks in patients with HB (George et al. 2017).
For HA, a few clinical trials have tested AAV vectors carrying a modified F8 gene with either a codon optimization (ubiquitination or specific amino acid sequence) or a deletion resulting in a FVIII without the B-domain (BDD-F8) but still compatible with a normal coagulant function and sufficient cassette packing (George and Fogarty 2016). BDD-F8 delivery by AAV5 has been approached under surveillance for possible immune reactions and controlled by prednisolone. This trial has shown relatively stable FVIII plasmatic activity (until 1 IU/dL) for three years in two patients after infusion (Pasi et al. 2020). Despite efforts to optimize the gene therapy through AAV vectors, previous exposure to AAV environmental serotypes generates neutralizing antibodies in 20-60% of the general population (Croteau et al. 2021). Therefore, many AAV-positive hemophilia patients must be excluded as candidates for therapy with AAV vectors.
Other strategies for the correction of a pathogenic variant are some enzyme systems. Zinc-finger proteins (ZFN) or transcription activator-like effector nuclease (TALEN) are designed to cleave specific sequences and subsequently generate knockouts by non-homologous end joining (NHEJ). Knocking with the wild-type sequence template can also be achieved by homologous direct recombination (HDR) using the natural DNA repair systems of cells.
Recently, the clustered regularly interspersed palindromic repeat (CRISPR) system, which works with a specific RNA guide sequence complementary to the target site, has emerged as a more specific and easier to use (in comparison to ZFN or TALEN) methodology. Thus, CRISPR technology is the perfect choice for generating monogenic disease models and correcting pathogenic variants (Ward and Walsh 2016).
CRISPR/Cas as a Gene-Edition Strategy for Monogenic Diseases
CRISPR system and CRISPR-associated proteins (Cas) constitute an adaptive immune system in Archaea and bacteria; it is composed of two classes, six types, and 33 subtypes of proteins with several functions. The class 1 system includes several Cas proteins to cleave the DNA while the class 2 system has a single, large, and multidomain binding Cas protein that can work as a class 1 complex (Makarova et al. 2020). Particularly, S. pyogenes Cas9 (type II from the class 2 CRISPR/Cas system) is the enzyme most widely used to modify eukaryotic genomes (Newsom et al. 2021). Cas9 works with the Watson-Crick complementarity principle between RNA and DNA; therefore, the use of single-guide RNA (sgRNA) to flank a 20-nucleotide complementary sequence is enough to generate a double-stranded break (DSB) and induce a NHEJ next to the protospacer adjacent motif (PAM) from the binding site (González-Romero et al. 2019). sgRNA and Cas9 complex can induce a DSB in any site-specific DNA target, demonstrating the wide spectrum of CRISPR/Cas9 system applications as a genome editing strategy on bacterial DNA (Jinek et al. 2012; Doudna and Charpentier 2014) or in different types of human cells (Mali et al. 2013; Jinek et al. 2013).
Cas9 and sgRNA can be introduced into target cells with several strategies like plasmid DNA, lentiviral vectors, mRNA, or pre-assembled ribonucleoprotein (RNP) complexes for in vitro, in vivo, and ex vivo approaches (Lino et al. 2018). RNP complex is one of the best options for clinical therapy because of its high efficiency and ephemeral action (low nuclease exposition on genome’s cells), a condition that decreases the risk of off-target effects (González-Romero et al. 2019). The risk of non-specific or off-target cleavage is about once per thousand cells or higher; therefore, the efficiency and safety of the CRISPR edition, as well as the potential risks of genotoxic non-specific cleavages, must be evaluated (Ward and Walsh 2016; González-Romero et al. 2019).
A possible solution to off-target risk is the Cas9 substitution by other Cas proteins (like Cas12a, also known as “Cpf1”) (Zetsche et al. 2015), or the use of nucleases guided by two different sgRNAs but targeting the same DNA locus, though in opposite senses to make the gene edition safer and more controlled (Wu et al. 2018). The main unknowns that should be clarified to implement the CRISPR/Cas9 system for gene edition of complex pathologies like cancer or autoimmune diseases are the minimum number of necessary edited cells to rescue the function and the immune response against the system (Shi et al. 2018; González-Romero et al. 2019).
CRISPR/Cas9 and Hemophilia
CRISPR/Cas9 offers great potential in research and translational studies in hematological diseases. Researchers use the system to generate cell cultures or experimental animal models with known pathogenic variants; however, the main goal is the ex vivo correction in the patient’s cells. The edition of a pathogenic variant on genomic DNA might be a definitive treatment through autologous transplants from patients to avoid the possible adverse effects as an immune system reaction (González-Romero et al. 2019). The CRISPR/Cas9 system has been used in hemophilia experimental in vitro, in vivo and in situ models, but no human clinical trial has been attempted yet (Guan et al. 2016; Croteau et al. 2021).
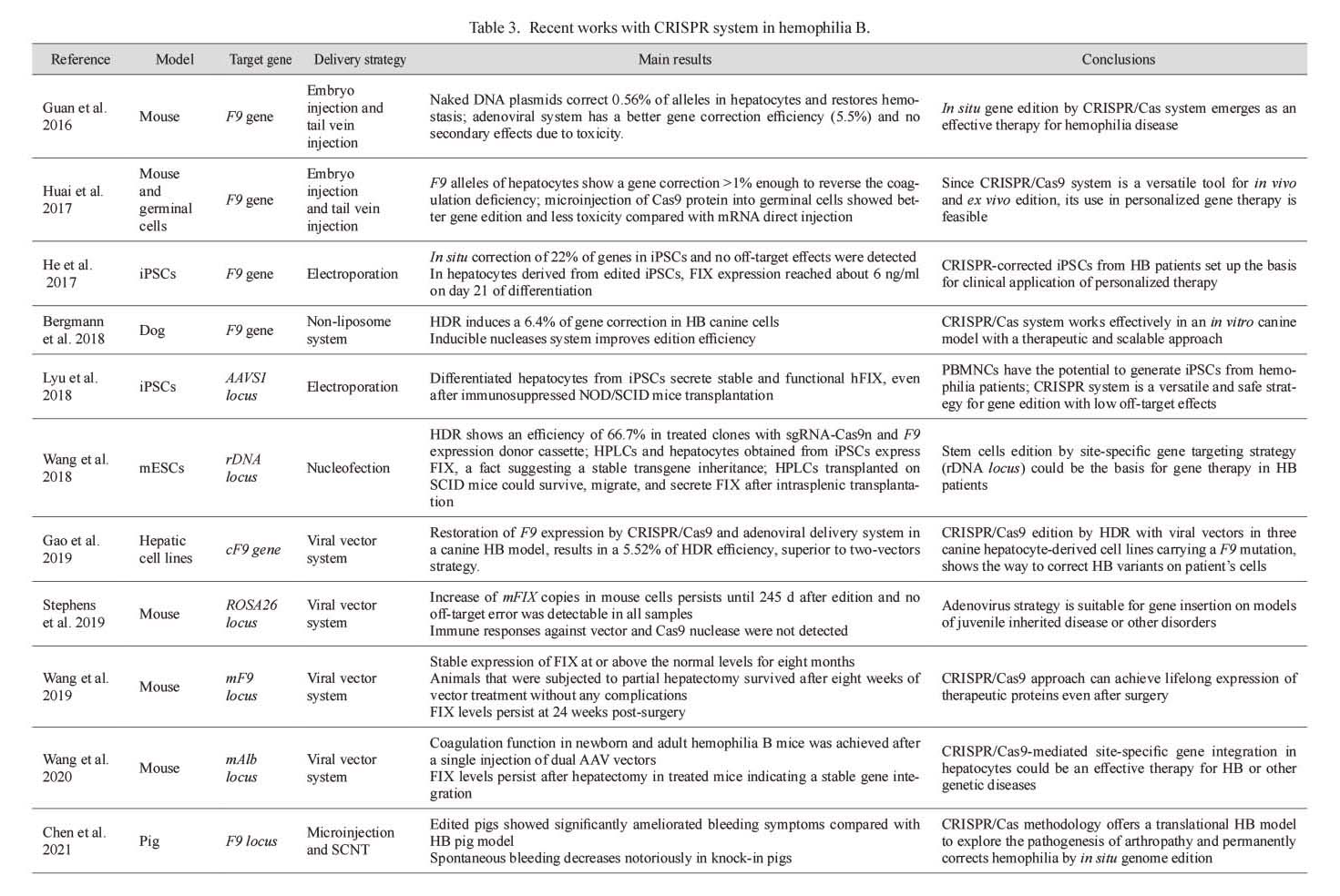
Particularly, induced pluripotent stem cells (iPSCs) and hepatic, endothelial, and platelet cells have been used to generate knock-out and knock-in models for HA therapy in which the CRISPR system and BDD-F8 modified protein could reverse the HA phenotype (Table 2). The CRISPR/Cas9 system has also been used to generate HB models in mice, dogs, and pigs in which plasma therapeutic values of the FIX protein are achieved after gene edition (Table 3).
In vivo gene edition trials for hemophilia are being developed despite the possible off-target effects of CRISPR technology. This is the most important concern when CRISPR is considered as therapy for hemophilia patients, especially in non-controlled in vivo gene edition; besides, the delivery strategy for some tissues may result in an immune response. Therefore, the CRISPR/Cas system in PWH would need constant and systemic monitoring of undesired off-target effects for years, compared to ex vivo editing, which is more simple and tissue-specific. Future safe clinical applications of the CRISPR system will require gene edition control tools such as turning the on/off switches of Cas activity according to some specific conditions, to avoid prolonged DNA rupture (Ernst et al. 2020).
However, the easy implementation and development of a CRISPR/Cas strategy for gene edition (with just one or two guide RNAs needed to flank a DNA locus) might allow for a specific and personalized gene therapy, regardless of the PWH’s pathogenic variant (Chen et al. 2019). Particularly, the CRISPR/Cas system in hemophilia remains a promising option because it is possible to edit different cell types that can produce active FVIII and FIX factors. CRISPR/Cas system has not yet been used on hemophilia patients, but a wide variety of pathologies like hereditary immune system disorders, congenital eye diseases, lipoprotein lipase deficiency, and genetically engineered T cells for cancer are some examples in which the ex vivo gene therapy with the CRISPR system works as an alternative to conventional pharmacotherapy (Odiba et al. 2021).
To date, CRISPR/Cas9 system is recognized as the most feasible tool for therapeutic gene edition; however, its use is limited because it is associated with a possible high frequency of off-target cleavages (Croteau et al. 2021). Technical limitations and long-term safety after gene edition are still to be overcome before the use of the CRISPR/Cas9 system evolves from a plausible promise to “the true hemophilia cure”. New experiments and long clinical trials are necessary to assess the risk-to-benefit ratio of CRISPR/Cas9 therapy before its direct use with hemophilia patients (González-Romero et al. 2019; Wang et al. 2019; Pipe and Selvaraj 2019).
Because the implicit scientific, ethical, and technological challenges, gene therapy requires more effort and larger clinical trials to facilitate its translation into clinical practice (Pipe and Selvaraj 2019). Recently published works on gene therapy for hemophilia depict an outlook of this strategy as a plausible treatment for different genetic diseases; in the future, it might be included by health systems as a definite cure for genetic disorders (Ernst et al. 2020).
Conclusions
The traditional replacement therapy for hemophilia has been substituted for novel treatments such as the bioengineered factor VII and IX molecules and non-factor treatment like Emicizumab antibody, which constitute efficient, safe, and long-lasting therapies that improve the quality of life of hemophilia patients more than ever.
Despite their amazing effectiveness, these therapies have limited coverture according to the half-life of recombinant proteins. This is overcome by gene therapy that potentially offers a definite cure through the correction of the pathogenic variants causing hemophilia.
Currently, gene therapy for hemophilia is more tangible due to advances and results in clinical trials with AAV vectors; however, in the better of future scenarios, viral vectors will be replaced by more secure and specific strategies like the CRISPR/Cas system. Any pathogenic variant that causes hemophilia could be corrected through ex vivo therapy in the patient’s cells, generating an individual and specific treatment for each hemophilia patient with the promise of a “real” long-term treatment without the continuous factor infusion or the risk of developing inhibitors.
CRISPR/Cas for gene therapy will soon be the first choice for hemophiliacs who are most severely affected by the disease. With the recent technical advances for safety optimization, gene transfer has matured as a real therapeutic option for hemophilia and other bleeding disorders, making it an individualized medicine approach of great interest for research, translational medicine, and the pharmaceutical industry.
Acknowledgments
We thank Dr. Horacio Rivera for his valuable revision of the manuscript.
Conflict of Interest
The authors declare no conflict of interest.
References
-
Asokan,
A.,
Schaffer,
D.V. &
Samulski,
R.J.
(2012) The AAV vector toolkit: poised at the clinical crossroads. Mol. Ther., 20, 699-708.
-
Bergmann,
T.,
Ehrke-Schulz,
E.,
Gao,
J.,
Schiwon,
M.,
Schildgen,
V.,
David,
S.,
Schildgen,
O. &
Ehrhardt,
A.
(2018) Designer nuclease-mediated gene correction via homology-directed repair in an in vitro model of canine hemophilia B. J. Gene Med., 20, e3020.
-
Blanchette,
V.S.,
Key,
N.S.,
Ljung,
L.R.,
Manco-Johnson,
M.J.,
van den Berg,
H.M. &
Srivastava,
A.;
Subcommittee on Factor VIII, Factor IX and Rare Coagulation Disorders of the Scientific and Standardization Committee of the International Society on Thrombosis and Hemostasis
(2014) Definitions in hemophilia: communication from the SSC of the ISTH. J. Thromb. Haemost., 12, 1935-1939.
-
Chen,
H.,
Shi,
M.,
Gilam,
A.,
Zheng,
Q.,
Zhang,
Y.,
Afrikanova,
I.,
Li,
J.,
Gluzman,
Z.,
Jiang,
R.,
Kong,
L.J. &
Chen-Tsai,
R.Y.
(2019) Hemophilia A ameliorated in mice by CRISPR-based in vivo genome editing of human Factor VIII. Sci. Rep., 9, 16838.
-
Chen,
J.,
An,
B.,
Yu,
B.,
Peng,
X.,
Yuan,
H.,
Yang,
Q.,
Chen,
X.,
Yu,
T.,
Wang,
L.,
Zhang,
X.,
Wang,
H.,
Zou,
X.,
Pang,
D.,
Ouyang,
H. &
Tang,
X.
(2021) CRISPR/Cas9-mediated knockin of human factor IX into swine factor IX locus effectively alleviates bleeding in hemophilia B pigs. Haematologica, 106, 829-837.
-
Collins,
P.,
Chalmers,
E.,
Chowdary,
P.,
Keeling,
D.,
Mathias,
M.,
O’Donnell,
J.,
Pasi,
K.J.,
Rangarajan,
S. &
Thomas,
A.
(2016) The use of enhanced half-life coagulation factor concentrates in routine clinical practice: guidance from UKHCDO. Haemophilia, 22, 487-498.
-
Croteau,
S.E.,
Wang,
M. &
Wheeler,
A.P.
(2021) 2021 clinical trials update: innovations in hemophilia therapy. Am. J. Hematol., 96, 128-144.
-
Doudna,
J.A. &
Charpentier,
E.
(2014) Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science, 346, 1258096.
-
Dumont,
J.A.,
Liu,
T.,
Low,
S.C.,
Zhang,
X.,
Kamphaus,
G.,
Sakorafas,
P.,
Fraley,
C.,
Drager,
D.,
Reidy,
T.,
McCue,
J.,
Franck,
H.W.,
Merricks,
E.P.,
Nichols,
T.C.,
Bitonti,
A.J.,
Pierce,
G.F.,
et al. (2012) Prolonged activity of a recombinant factor VIII-Fc fusion protein in hemophilia A mice and dogs. Blood, 119, 3024-3030.
-
Ernst,
M.P.T.,
Broeders,
M.,
Herrero-Hernandez,
P.,
Oussoren,
E.,
van der Ploeg,
A.T. &
Pijnappel,
W.
(2020) Ready for repair? Gene editing enters the clinic for the treatment of human disease. Mol. Ther. Methods Clin. Dev., 18, 532-557.
-
Escobar,
M.,
Santagostino,
E.,
Mancuso,
M.E.,
Coppens,
M.,
Balasa,
V.,
Taylor,
J.A.,
Iorio,
A. &
Negrier,
C.
(2019) Switching patients in the age of long-acting recombinant products? Expert Rev. Hematol., 12, 1-13.
-
Evens,
H.,
Chuah,
M.K. &
VandenDriessche,
T.
(2018) Haemophilia gene therapy: from trailblazer to gamechanger . Haemophilia, 24 Suppl 6, 50-59.
-
Gao,
J.,
Bergmann,
T.,
Zhang,
W.,
Schiwon,
M.,
Ehrke-Schulz,
E. &
Ehrhardt,
A.
(2019) Viral vector-based delivery of CRISPR/Cas9 and donor DNA for homology-directed repair in an in vitro model for canine hemophilia B. Mol. Ther. Nucleic Acids, 14, 364-376.
-
George,
L.A. &
Fogarty,
P.F.
(2016) Gene therapy for hemophilia: past, present and future. Semin. Hematol., 53, 46-54.
-
George,
L.A.,
Sullivan,
S.K.,
Giermasz,
A.,
Rasko,
J.E.J.,
Samelson-Jones,
B.J.,
Ducore,
J.,
Cuker,
A.,
Sullivan,
L.M.,
Majumdar,
S.,
Teitel,
J.,
McGuinn,
C.E.,
Ragni,
M.V.,
Luk,
A.Y.,
Hui,
D.,
Wright,
J.F.,
et al. (2017) Hemophilia B gene therapy with a high-specific-activity factor IX variant. N. Engl. J. Med., 377, 2215-2227.
-
González-Romero,
E.,
Martinez-Valiente,
C.,
Garcia-Ruiz,
C.,
Vazquez-Manrique,
R.P.,
Cervera,
J. &
Sanjuan-Pla,
A.
(2019) CRISPR to fix bad blood: a new tool in basic and clinical hematology. Haematologica, 104, 881-893.
-
Guan,
Y.,
Ma,
Y.,
Li,
Q.,
Sun,
Z.,
Ma,
L.,
Wu,
L.,
Wang,
L.,
Zeng,
L.,
Shao,
Y.,
Chen,
Y.,
Ma,
N.,
Lu,
W.,
Hu,
K.,
Han,
H.,
Yu,
Y.,
et al. (2016) CRISPR/Cas9-mediated somatic correction of a novel coagulator factor IX gene mutation ameliorates hemophilia in mouse. EMBO Mol. Med., 8, 477-488.
-
Guo,
X.L.,
Chung,
T.H.,
Qin,
Y.,
Zheng,
J.,
Zheng,
H.,
Sheng,
L.,
Wynn,
T. &
Chang,
L.J.
(2019) Hemophilia gene therapy: new development from bench to bed side. Curr. Gene Ther., 19, 264-273.
-
Hartmann,
R.,
Feenstra,
T.,
Valentino,
L.,
Dockal,
M. &
Scheiflinger,
F.
(2018) In vitro studies show synergistic effects of a procoagulant bispecific antibody and bypassing agents. J. Thromb. Haemost., 16, 1580-1591.
-
He,
Q.,
Wang,
H.H.,
Cheng,
T.,
Yuan,
W.P.,
Ma,
Y.P.,
Jiang,
Y.P. &
Ren,
Z.H.
(2017) Genetic correction and hepatic differentiation of hemophilia B-specific human induced pluripotent stem cells. Chin. Med. Sci. J., 32, 135-144.
-
Hermans,
C. &
Dolan,
G.
(2020) Pharmacokinetics in routine haemophilia clinical practice: rationale and modalities-a practical review. Ther. Adv. Hematol., 11, 2040620720966888.
-
High,
K.H.,
Nathwani,
A.,
Spencer,
T. &
Lillicrap,
D.
(2014) Current status of haemophilia gene therapy. Haemophilia, 20 Suppl 4, 43-49.
-
Hu,
Z.,
Zhou,
M.,
Wu,
Y.,
Li,
Z.,
Liu,
X.,
Wu,
L. &
Liang,
D.
(2019) ssODN-mediated in-frame deletion with CRISPR/Cas9 restores FVIII function in hemophilia A-patient-derived iPSCs and ECs. Mol. Ther. Nucleic Acids, 17, 198-209.
-
Huai,
C.,
Jia,
C.,
Sun,
R.,
Xu,
P.,
Min,
T.,
Wang,
Q.,
Zheng,
C.,
Chen,
H. &
Lu,
D.
(2017) CRISPR/Cas9-mediated somatic and germline gene correction to restore hemostasis in hemophilia B mice. Hum. Genet., 136, 875-883.
-
Iorio,
A.,
Stonebraker,
J.S.,
Chambost,
H.,
Makris,
M.,
Coffin,
D.,
Herr,
C. &
Germini,
F.;
Data and Demographics Committee of the World Federation of Hemophilia
(2019) Establishing the prevalence and prevalence at birth of hemophilia in males: a meta-analytic approach using national registries. Ann. Intern. Med., 171, 540-546.
-
Ivens,
I.A.,
Baumann,
A.,
McDonald,
T.A.,
Humphries,
T.J.,
Michaels,
L.A. &
Mathew,
P.
(2013) PEGylated therapeutic proteins for haemophilia treatment: a review for haemophilia caregivers. Haemophilia, 19, 11-20.
-
Jinek,
M.,
Chylinski,
K.,
Fonfara,
I.,
Hauer,
M.,
Doudna,
J.A. &
Charpentier,
E.
(2012) A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science, 337, 816-821.
-
Jinek,
M.,
East,
A.,
Cheng,
A.,
Lin,
S.,
Ma,
E. &
Doudna,
J.
(2013) RNA-programmed genome editing in human cells. Elife, 2, e00471.
-
Kitazawa,
T.,
Igawa,
T.,
Sampei,
Z.,
Muto,
A.,
Kojima,
T.,
Soeda,
T.,
Yoshihashi,
K.,
Okuyama-Nishida,
Y.,
Saito,
H.,
Tsunoda,
H.,
Suzuki,
T.,
Adachi,
H.,
Miyazaki,
T.,
Ishii,
S.,
Kamata-Sakurai,
M.,
et al. (2012) A bispecific antibody to factors IXa and X restores factor VIII hemostatic activity in a hemophilia A model. Nat. Med., 18, 1570-1574.
-
Kleiboer,
B.,
Nielsen,
B.,
Ma,
A.D.,
Abajas,
Y.,
Monroe,
D.M. &
Key,
N.S.
(2020) Excessive breakthrough bleeding in haemophilia B patients on factor IX-albumin fusion protein prophylactic therapy: a single centre case series. Haemophilia, 26, e23-e25.
-
Kotin,
R.M.,
Linden,
R.M. &
Berns,
K.I.
(1992) Characterization of a preferred site on human chromosome 19q for integration of adeno-associated virus DNA by non-homologous recombination. EMBO J., 11, 5071-5078.
-
Lieuw,
K.
(2017) Many factor VIII products available in the treatment of hemophilia A: an embarrassment of riches? J. Blood Med., 8, 67-73.
-
Lino,
C.A.,
Harper,
J.C.,
Carney,
J.P. &
Timlin,
J.A.
(2018) Delivering CRISPR: a review of the challenges and approaches. Drug Deliv., 25, 1234-1257.
-
Ljung,
R.C.R.
(2018) Prevention and management of bleeding episodes in children with hemophilia. Paediatr. Drugs, 20, 455-464.
-
Lyu,
C.,
Shen,
J.,
Wang,
R.,
Gu,
H.,
Zhang,
J.,
Xue,
F.,
Liu,
X.,
Liu,
W.,
Fu,
R.,
Zhang,
L.,
Li,
H.,
Zhang,
X.,
Cheng,
T.,
Yang,
R. &
Zhang,
L.
(2018) Targeted genome engineering in human induced pluripotent stem cells from patients with hemophilia B using the CRISPR-Cas9 system. Stem Cell. Res. Ther., 9, 92.
-
Mahlangu,
J.,
Oldenburg,
J.,
Paz-Priel,
I.,
Negrier,
C.,
Niggli,
M.,
Mancuso,
M.E.,
Schmitt,
C.,
Jimenez-Yuste,
V.,
Kempton,
C.,
Dhalluin,
C.,
Callaghan,
M.U.,
Bujan,
W.,
Shima,
M.,
Adamkewicz,
J.I.,
Asikanius,
E.,
et al. (2018) Emicizumab prophylaxis in patients who have hemophilia A without inhibitors. N. Engl. J. Med., 379, 811-822.
-
Mahlangu,
J.,
Powell,
J.S.,
Ragni,
M.V.,
Chowdary,
P.,
Josephson,
N.C.,
Pabinger,
I.,
Hanabusa,
H.,
Gupta,
N.,
Kulkarni,
R.,
Fogarty,
P.,
Perry,
D.,
Shapiro,
A.,
Pasi,
K.J.,
Apte,
S.,
Nestorov,
I.,
et al. (2014) Phase 3 study of recombinant factor VIII Fc fusion protein in severe hemophilia A. Blood, 123, 317-325.
-
Makarova,
K.S.,
Wolf,
Y.I.,
Iranzo,
J.,
Shmakov,
S.A.,
Alkhnbashi,
O.S.,
Brouns,
S.J.J.,
Charpentier,
E.,
Cheng,
D.,
Haft,
D.H.,
Horvath,
P.,
Moineau,
S.,
Mojica,
F.J.M.,
Scott,
D.,
Shah,
S.A.,
Siksnys,
V.,
et al. (2020) Evolutionary classification of CRISPR-Cas systems: a burst of class 2 and derived variants. Nat. Rev. Microbiol., 18, 67-83.
-
Malec,
L.M.,
Croteau,
S.E.,
Callaghan,
M.U. &
Sidonio,
R.F. Jr.
(2020) Spontaneous bleeding and poor bleeding response with extended half-life factor IX products: a survey of select US haemophilia treatment centres. Haemophilia, 26, e128-e129.
-
Mali,
P.,
Yang,
L.,
Esvelt,
K.M.,
Aach,
J.,
Guell,
M.,
DiCarlo,
J.E.,
Norville,
J.E. &
Church,
G.M.
(2013) RNA-guided human genome engineering via Cas9. Science, 339, 823-826.
-
Mancuso,
M.E. &
Santagostino,
E.
(2017) Outcome of clinical trials with new extended half-life FVIII/IX concentrates. J. Clin. Med., 6, 39.
-
Mannucci,
P.M.
(2020) Hemophilia therapy: the future has begun. Haematologica, 105, 545-553.
-
Meeks,
S.L. &
Lacroix-Desmazes,
S.
(2020) Emerging benefits of Fc fusion technology in the context of recombinant factor VIII replacement therapy. Haemophilia, 26, 958-965.
-
Monahan,
P.E.
(2015) Gene therapy in an era of emerging treatment options for hemophilia B. J. Thromb. Haemost., 13 Suppl 1, S151-160.
-
Morfini,
M. &
Marchesini,
E.
(2020) The availability of new drugs for hemophilia treatment. Expert Rev. Clin. Pharmacol., 13, 721-738.
-
Morfini,
M. &
Rapisarda,
C.A.P.
(2019) Safety of recombinant coagulation factors in treating hemophilia. Expert Opin. Drug Saf., 18, 75-85.
-
Nathwani,
A.C.,
Reiss,
U.M.,
Tuddenham,
E.G.,
Rosales,
C.,
Chowdary,
P.,
McIntosh,
J.,
Della Peruta,
M.,
Lheriteau,
E.,
Patel,
N.,
Raj,
D.,
Riddell,
A.,
Pie,
J.,
Rangarajan,
S.,
Bevan,
D.,
Recht,
M.,
et al. (2014) Long-term safety and efficacy of factor IX gene therapy in hemophilia B. N. Engl. J. Med., 371, 1994-2004.
-
Negrier,
C.
(2016) Entering new areas in known fields: recombinant fusion protein linking recombinant factor VIIa with recombinant albumin (rVIIa-FP) - advancing the journey. Thromb. Res., 141 Suppl 3, S9-S12.
-
Newsom,
S.,
Parameshwaran,
H.P.,
Martin,
L. &
Rajan,
R.
(2021) The CRISPR-Cas mechanism for adaptive immunity and alternate bacterial functions fuels diverse biotechnologies. Front. Cell. Infect. Microbiol., 10, 619763.
-
Odiba,
A.S.,
Okoro,
N.O.,
Durojaye,
O.A. &
Wu,
Y.
(2021) Gene therapy in PIDs, hemoglobin, ocular, neurodegenerative, and hemophilia B disorders. Open Life Sci., 16, 431-441.
-
Oldenburg,
J.,
Mahlangu,
J.N.,
Kim,
B.,
Schmitt,
C.,
Callaghan,
M.U.,
Young,
G.,
Santagostino,
E.,
Kruse-Jarres,
R.,
Negrier,
C.,
Kessler,
C.,
Valente,
N.,
Asikanius,
E.,
Levy,
G.G.,
Windyga,
J. &
Shima,
M.
(2017) Emicizumab prophylaxis in hemophilia A with inhibitors. N. Engl. J. Med., 377, 809-818.
-
Olmedillas López,
S.,
Garcia-Arranz,
M.,
Garcia-Olmo,
D. &
Liras,
A.
(2016) Preliminary study on non-viral transfection of F9 (factor IX) gene by nucleofection in human adipose-derived mesenchymal stem cells. PeerJ, 4, e1907.
-
Park,
C.Y.,
Kim,
D.H.,
Son,
J.S.,
Sung,
J.J.,
Lee,
J.,
Bae,
S.,
Kim,
J.H.,
Kim,
D.W. &
Kim,
J.S.
(2015) Functional correction of large factor VIII gene chromosomal inversions in hemophilia A patient-derived iPSCs using CRISPR-Cas9. Cell Stem Cell, 17, 213-220.
-
Park,
C.Y.,
Sung,
J.J.,
Cho,
S.R.,
Kim,
J. &
Kim,
D.W.
(2019) Universal correction of blood coagulation factor VIII in patient-derived induced pluripotent stem cells using CRISPR/Cas9. Stem Cell Reports, 12, 1242-1249.
-
Pasi,
K.J.,
Rangarajan,
S.,
Mitchell,
N.,
Lester,
W.,
Symington,
E.,
Madan,
B.,
Laffan,
M.,
Russell,
C.B.,
Li,
M.,
Pierce,
G.F. &
Wong,
W.Y.
(2020) Multiyear follow-up of AAV5-hFVIII-SQ gene therapy for hemophilia A. N. Engl. J. Med., 382, 29-40.
-
Pignani,
S.,
Zappaterra,
F.,
Barbon,
E.,
Follenzi,
A.,
Bovolenta,
M.,
Bernardi,
F.,
Branchini,
A. &
Pinotti,
M.
(2019) Tailoring the CRISPR system to transactivate coagulation gene promoters in normal and mutated contexts. Biochim. Biophys. Acta Gene. Regul. Mech., 1862, 619-624.
-
Pipe,
S.W. &
Selvaraj,
S.R.
(2019) Gene editing in hemophilia: a “CRISPR” choice? Blood, 133, 2733-2734.
-
Powell,
J.S.,
Josephson,
N.C.,
Quon,
D.,
Ragni,
M.V.,
Cheng,
G.,
Li,
E.,
Jiang,
H.,
Li,
L.,
Dumont,
J.A.,
Goyal,
J.,
Zhang,
X.,
Sommer,
J.,
McCue,
J.,
Barbetti,
M.,
Luk,
A.,
et al. (2012) Safety and prolonged activity of recombinant factor VIII Fc fusion protein in hemophilia A patients. Blood, 119, 3031-3037.
-
Preijers,
T.,
Bukkems,
L.,
van Spengler,
M.,
Leebeek,
F.,
Cnossen,
M. &
Mathot,
R.
(2021) In silico comparison of pharmacokinetic properties of three extended half-life factor IX concentrates. Eur. J. Clin. Pharmacol., 77, 1193-1200.
-
Santagostino,
E.,
Negrier,
C.,
Klamroth,
R.,
Tiede,
A.,
Pabinger-Fasching,
I.,
Voigt,
C.,
Jacobs,
I. &
Morfini,
M.
(2012) Safety and pharmacokinetics of a novel recombinant fusion protein linking coagulation factor IX with albumin (rIX-FP) in hemophilia B patients. Blood, 120, 2405-2411.
-
Schulte,
S.
(2013) Innovative coagulation factors: albumin fusion technology and recombinant single-chain factor VIII. Thromb. Res., 131 Suppl 2, S2-6.
-
Shapiro,
A.,
Chaudhury,
A.,
Wang,
M.,
Escobar,
M.,
Tsao,
E.,
Barnowski,
C.,
Feng,
J.,
Jain,
N. &
Quon,
D.V.
(2020) Real-world data demonstrate improved bleed control and extended dosing intervals for patients with haemophilia B after switching to recombinant factor IX Fc fusion protein (rFIXFc) for up to 5 years. Haemophilia, 26, 975-983.
-
Shi,
Q.,
Mattson,
J.G.,
Fahs,
S.A.,
Geurts,
A.M.,
Weiler,
H. &
Montgomery,
R.R.
(2020) The severe spontaneous bleeding phenotype in a novel hemophilia A rat model is rescued by platelet FVIII expression. Blood Adv., 4, 55-65.
-
Shi,
X.,
Kitano,
A.,
Jiang,
Y.,
Luu,
V.,
Hoegenauer,
K.A. &
Nakada,
D.
(2018) Clonal expansion and myeloid leukemia progression modeled by multiplex gene editing of murine hematopoietic progenitor cells. Exp. Hematol., 64, 33-44 e35.
-
Stephens,
C.J.,
Lauron,
E.J.,
Kashentseva,
E.,
Lu,
Z.H.,
Yokoyama,
W.M. &
Curiel,
D.T.
(2019) Long-term correction of hemophilia B using adenoviral delivery of CRISPR/Cas9. J. Control. Release, 298, 128-141.
-
Sung,
J.J.,
Park,
C.Y.,
Leem,
J.W.,
Cho,
M.S. &
Kim,
D.W.
(2019) Restoration of FVIII expression by targeted gene insertion in the FVIII locus in hemophilia A patient-derived iPSCs. Exp. Mol. Med., 51, 1-9.
-
Sung,
J.J.,
Park,
S.,
Choi,
S.H.,
Kim,
J.,
Cho,
M.S. &
Kim,
D.W.
(2020) Generation of a gene edited hemophilia A patient-derived iPSC cell line, YCMi001-B-1, by targeted insertion of coagulation factor FVIII using CRISPR/Cas9. Stem Cell Res., 48, 101948.
-
Swierczewska,
M.,
Lee,
K.C. &
Lee,
S.
(2015) What is the future of PEGylated therapies? Expert Opin. Emerg. Drugs, 20, 531-536.
-
VandenDriessche,
T. &
Chuah,
M.K.
(2017) Hemophilia gene therapy: ready for prime time? Hum. Gene Ther., 28, 1013-1023.
-
Wang,
L.,
Yang,
Y.,
Breton,
C.A.,
White,
J.,
Zhang,
J.,
Che,
Y.,
Saveliev,
A.,
McMenamin,
D.,
He,
Z.,
Latshaw,
C.,
Li,
M. &
Wilson,
J.M.
(2019) CRISPR/Cas9-mediated in vivo gene targeting corrects hemostasis in newborn and adult factor IX-knockout mice. Blood, 133, 2745-2752.
-
Wang,
Q.,
Zhong,
X.,
Li,
Q.,
Su,
J.,
Liu,
Y.,
Mo,
L.,
Deng,
H. &
Yang,
Y.
(2020) CRISPR-Cas9-mediated in vivo gene integration at the albumin locus recovers hemostasis in neonatal and adult hemophilia B mice. Mol. Ther. Methods Clin. Dev., 18, 520-531.
-
Wang,
Y.,
Zhao,
J.,
Duan,
N.,
Liu,
W.,
Zhang,
Y.,
Zhou,
M.,
Hu,
Z.,
Feng,
M.,
Liu,
X.,
Wu,
L.,
Li,
Z. &
Liang,
D.
(2018) Paired CRISPR/Cas9 nickases mediate efficient site-specific integration of F9 into rDNA locus of mouse ESCs. Int. J. Mol. Sci., 19, 3035.
-
Ward,
P. &
Walsh,
C.E.
(2016) Current and future prospects for hemophilia gene therapy. Expert Rev. Hematol., 9, 649-659.
-
Weyand,
A.C. &
Pipe,
S.W.
(2019) New therapies for hemophilia. Blood, 133, 389-398.
-
World Federation of Hemophilia
(2019) Report on the Annual Global Survey 2019. https://elearning.wfh.org/resource/report-on-the-annual-global-survey-2019/ [Accessed: September 13, 2021].
-
Wu,
W.Y.,
Lebbink,
J.H.G.,
Kanaar,
R.,
Geijsen,
N. &
van der Oost,
J.
(2018) Genome editing by natural and engineered CRISPR-associated nucleases. Nat. Chem. Biol., 14, 642-651.
-
Young,
G.,
Liesner,
R.,
Chang,
T.,
Sidonio,
R.,
Oldenburg,
J.,
Jimenez-Yuste,
V.,
Mahlangu,
J.,
Kruse-Jarres,
R.,
Wang,
M.,
Uguen,
M.,
Doral,
M.Y.,
Wright,
L.Y.,
Schmitt,
C.,
Levy,
G.G.,
Shima,
M.,
et al. (2019) A multicenter, open-label phase 3 study of emicizumab prophylaxis in children with hemophilia A with inhibitors. Blood, 134, 2127-2138.
-
Zetsche,
B.,
Gootenberg,
J.S.,
Abudayyeh,
O.O.,
Slaymaker,
I.M.,
Makarova,
K.S.,
Essletzbichler,
P.,
Volz,
S.E.,
Joung,
J.,
van der Oost,
J.,
Regev,
A.,
Koonin,
E.V. &
Zhang,
F.
(2015) Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell, 163, 759-771.
-
Zhang,
J.P.,
Cheng,
X.X.,
Zhao,
M.,
Li,
G.H.,
Xu,
J.,
Zhang,
F.,
Yin,
M.D.,
Meng,
F.Y.,
Dai,
X.Y.,
Fu,
Y.W.,
Yang,
Z.X.,
Arakaki,
C.,
Su,
R.J.,
Wen,
W.,
Wang,
W.T.,
et al. (2019) Curing hemophilia A by NHEJ-mediated ectopic F8 insertion in the mouse. Genome Biol., 20, 276.