Abstract
Myositis-specific autoantibodies are relevant factors that define the disease phenotype of dermatomyositis (DM). Anti-Mi-2 antibody-positive DM patients may present with the typical skin lesions and prominent myositis. On the other hand, adult DM patients with anti-TIF-γ antibody seem to be associated with internal malignancy. Here, we report a rare case of juvenile dermatomyositis (JDM) exhibiting anti-Mi-2 and anti-transcriptional intermediary factor-1 gamma (TIF1-γ) antibodies, with no internal malignancy. A 16-year-old female Japanese patient under treatment with a 2-year history of chronic eczematous lesions was admitted to our department with elevated levels of muscle enzymes. Characteristic skin changes, such as Gottron’s papules of the hand, heliotrope rash of the eyelids, and poikiloderma-like legions and diffuse pigmentation on the back, were observed. Histologically, the patient’s skin was characterized by the presence of lymphocytic vascular inflammation and endothelial swelling, which are consistent with DM. Severe symmetric proximal muscle weakness, elevated serum muscle enzymes and the presence of anti-TIF1-γ and Mi-2 antibodies were noted. The diagnosis of JDM was made according to the European League Against Rheumatism (EULAR) diagnostic criteria. A high dose of corticosteroids and following intravenous cyclophosphamide treatment (750 mg three times) resulted in an improvement in clinical manifestations and functional outcomes, and recurrence did not occur. Estimation of autoantibodies may serve as an ancillary tool in delineating and defining distinct clinical phenotypes in JDM.
Introduction
Dermatomyositis (DM) is defined as a heterogeneous disorder that can occur in both adults and juveniles, and it has various manifestations, including myositis, dermatitis, and interstitial lung disease (ILD) (Tansley et al. 2013). Juvenile dermatomyositis (JDM) is a heterogeneous type of inflammatory myopathy, and dermatitis is mainly the first manifestation of the disease (Pagnini et al. 2017). The cutaneous manifestations of JDM are characterized by photosensitive rashes, the V-sign or shawl-sign rashes in addition to Gottron’s papules or heliotrope rash (Rider and Nistala 2016). Various myositis-specific autoantibodies (MSAs) have been reported as specific disease markers of DM (Fujimoto et al. 2016b). For example, anti-Mi-2 antibody-positive DM patients show typical skin lesions and myositis, but these patients rarely exhibit internal malignancy or ILD (Ghirardello et al. 2005). On the other hand, anti-transcriptional intermediary factor-1 gamma (TIF1-γ) antibodies have been detected in 22%-33% of DM patients, and the presence of these autoantibodies is a strong predictor of malignancy in adult DM (Stuhlmuller et al. 2019). A high frequency of heliotrope rash, V-sign, and Gottron’s papules and poikiloderma has been associated with DM patients with anti-TIF1-γ antibodies (Kotobuki et al. 2021). Herein, we report a patient with JDM exhibiting with both anti-Mi-2 and anti-TIF1-γ antibodies without ILD or internal malignancy.
Case Presentation
A 16-year-old Japanese female was admitted to our hospital for skin rash, left elbow joint pain, and bilateral thigh pain. The patient had no notable medical history. She presented with diffuse pruritic skin rash on the neck and elbows for 2 years prior to admission to our hospital. Eight months prior to admission, she developed worsening eczematous erythematous papules throughout her body and pigmentation appeared bilaterally on the upper limbs. Six months prior to admission, she visited her local dermatologist and was diagnosed with atopic dermatitis. She had no family history of atopic dermatitis and no complications such as bronchial asthma or allergic rhinitis. It was not clear whether dermatomyositis-specific antibody titers were measured during treatment period of her eczema at the nearby clinic. Steroid ointment was then prescribed, but the pigmentation did not improve and had instead spread to the whole body.
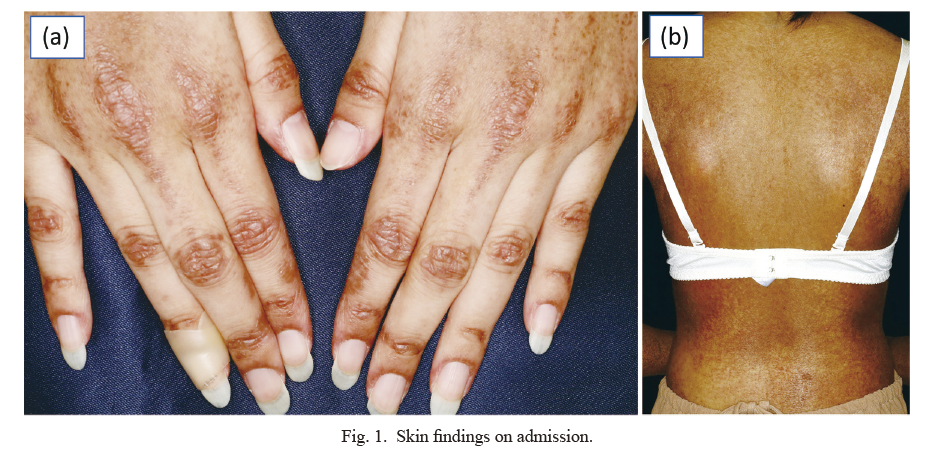
One month prior to admission, she became aware of bilateral thigh pain and elbow joint pain. The patient visited a local orthopedic clinic. The patient had unexplained muscle and soft tissue pain and was referred to our hospital. Physical examination revealed black pigmentation all over the body and a rash with scales on the extensor surfaces of the extremities and head. The fingers showed Gottron’s sign, and poikiloderma-like lesions and diffuse pigmentation on the back were observed (Fig. 1). Tenderness was noted in the bilateral elbow joints, and automatic and grasp pain was observed in the biceps and triceps brachii and bilateral quadriceps femoris. Manual muscle strength test showed muscle weakness with proximal muscle: iliopsoas 4/4, quadriceps 4/4, and hamstrings 4/4. The laboratory findings (Table 1) revealed elevated serum levels of creatine kinase at 16,499 U/l (reference range 41 to 153), lactate dehydrogenase at 1,620 U/l (reference range 124 to 222), C-reactive protein at 0.81 mg/dl (reference range up to 0.30), and aldolase at 158.6 U/l (reference range up to 7.5 U/l). Her liver enzymes were observed to be elevated, aspartate transaminase (412 U/l, reference range up to 30 U/l) and alanine transaminase (203 U/l, reference range up to 23 U/l). Her serum IgE was elevated at 9,096 IU/mL (reference range up to 169 IU/mL). Antinuclear antibody was positive (titer 1:1,280, reference range up to 1:160). Anti-aminoacyl transfer RNA synthetase (ARS) and anti-Jo-1 antibodies, which are specific antibodies for DM, were negative. However, anti-TIF1-γ and anti-Mi-2 antibodies were positive, at titers of 48.0 index (reference range up to 32.0 index) and over 150 index (reference range up to 53.0 index), respectively. JDM was suspected, and the patient was then admitted to our department for further examination and treatment.
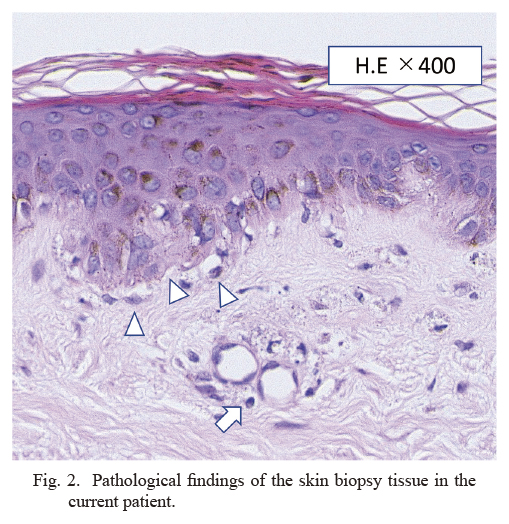
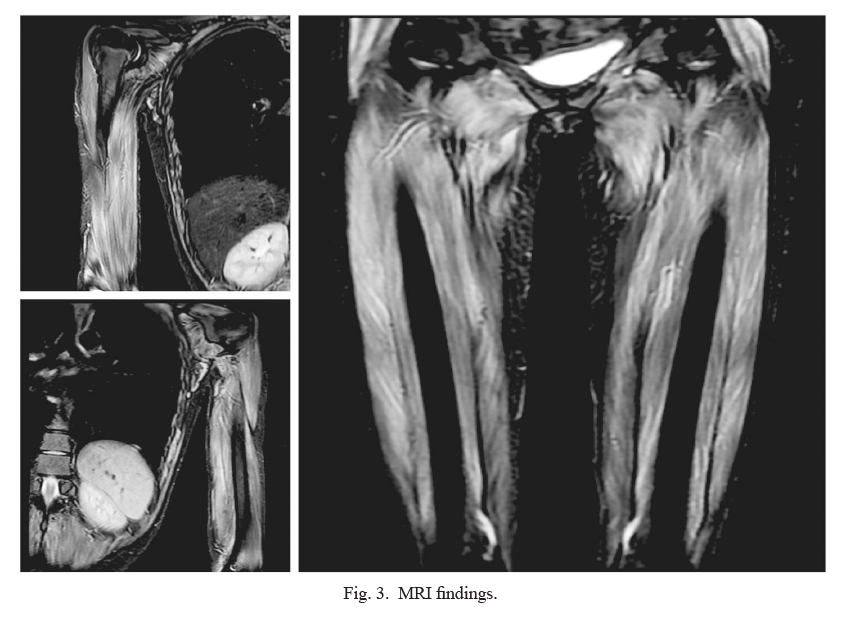
On admission, her temperature was 37.5°C, heart rate was 130 beats/min, and blood pressure was 115/76 mmHg. Blood culture tests were negative. A skin biopsy was performed to confirm the diagnosis. The specimen showed dyskeratotic cells in the epidermis and lymphocytic infiltration of the perivascular area within the dermis, findings consistent with DM (Fig. 2). Additionally, magnetic resonance imaging (MRI) of the bilateral upper and lower limbs showed high signal on short inversion time inversion recovery (STIR) that confirmed the findings of myositis (Fig. 3). Contrast-enhanced computed tomography (CECT) showed no findings suggestive of infection. Needle electromyography showed myogenic changes. According to the European League Against Rheumatism (EULAR)/the American College of Rheumatology (ACR) classification criteria for idiopathic inflammatory myopathies (IIMs), she had symmetrical muscle weakness of the proximal upper and lower limb muscles, Gottoron’s papules, Gottoron’s sign, and elevated serum levels of creatine kinase (CK). The patient was 16 years old; therefore, she was diagnosed with JDM. Tumor markers (CEA, CA19-9, and CA-125) were negative, and CECT showed no findings that indicated malignancy.
The clinical course is summarized in Fig. 4. Initially, the patient was started on prednisolone at 30 mg/day (1.0 mg per kg of body weight). However, due to symptoms of myalgia and a lack of improvement in myogenic enzymes in the biochemical tests, the patient received pulse therapy with corticosteroids [1,000 mg/day methylprednisolone intravenously for 3 days (days 6, 7, and 8), followed by 60 mg/day of oral prednisolone]. Then, 3.0 mg/day oral tacrolimus was started on the 12th day, and 750 mg intravenous cyclophosphamide (IV-CY) once every 4 weeks was administered starting on the 20th day. After the start of treatment, the generalized myalgia, skin rash, and muscle weakness were noted to improve. The levels of myogenic enzymes and inflammatory markers decreased. Prednisolone was gradually reduced, and the patient was discharged on the 51st day. Currently, she continues to attend the outpatient clinic, and IV-CY has been performed three times, tacrolimus has been reduced to 3.0 mg/day, and prednisolone has been reduced to 12.5 mg/day, but her symptoms have not flared up.
Informed consent was obtained from the patient. Because of a case report of single patient, ethical approval was waived for institutional review board in Fukushima Medical University.
Discussion
Anti-TIF1-γ antibodies are frequently associated with increased risk of cancer in adult patients with DM (Shimizu et al. 2020). On the other hand, anti-TIF1-γ antibody-positive JDM patients are rarely associated with cancer (Morris and Dare 2010). Additionally, juvenile and adult patients with anti-TIF1-γ autoantibodies tend to have a typical, severe skin rash, including heliotrope rash, the V-neck sign, the shawl sign, and Gottron’s papules with a chronic clinical course (Kotobuki et al. 2021). The majority of patients positive for anti-Mi-2 antibodies present with a DM phenotype with typical cutaneous manifestations, including heliotrope rash, Gottron’s sign, periungual erythema, and truncal erythema including the V-sign and the shawl sign, with prominent muscle weakness, a better therapeutic response, and less frequent cancer (Ogawa-Momohara et al. 2019). Additionally, anti-Mi-2 antibody-positive patients have been determined to have a low risk of developing clinically relevant ILD (Liang et al. 2020). Although these MSAs have been considered to be mutually exclusive, the present case demonstrates that this anti-TIF1 antibody-positive patient has classical DM without cancer and be associated with anti-Mi-2 antibody seropositivity. Anti-p155/140 antibodies have been found to be serological markers of cancer-associated DM (Kaji et al. 2007). The anti-p155 autoantibody has been detected against a 155-kd protein identified as TIF1-γ and p140 is identical to TIF1-α (Fujimoto et al. 2012). Recent studies showed that anti-TIF1-γ antibodies are detected in JDM patients (Rider et al. 2013). By contrast, anti-Mi-2 antibodies were detected less frequently in JDM patients compared to adult DM patients (Feldman et al. 1996). Anti-Mi-2 antibodies mainly react to Mi-2β, which is a component of the nucleosome-remodeling deacetylase complex (Fujimoto et al. 2016a). Anti-Mi-2 antibody may cross-react with TIF1-α protein via its plant homeodomain, which is highly homologous to that of the Mi-2β protein (Fujimoto et al. 2016a). Fujimoto et al. (2016a) demonstrated that anti-TIF1-α/γ double positivity was more common than anti-TIF1-α-positive or anti-TIF1-γ-negative sera. Although anti-TIF1-α antibodies were not evaluated in the present case, it is possible that anti-TIF1-γ antibodies and anti-Mi-2 antibodies can coexist in a subset of DM patients with anti-TIF1-α/γ double positivity due to the cross-reactivity of anti-TIF1-α antibodies. Indeed, a case of JDM with both anti-TIF1-γ and anti-Mi-2 antibodies has been reported (Takezaki et al. 2020). According to the report, our case had similar symptoms, typical severe skin rash, no malignancy, as demonstrated previously (Takezaki et al. 2020).
Expression of Mi-2 was found to be markedly upregulated during muscle regeneration in a mouse model of muscle injury and repair (Mammen et al. 2009). In the skin, Mi-2 has been shown to be essential for the repair of the basal epidermis, and UV radiation induces a high expression of Mi-2 in keratinocytes (Burd et al. 2008). These findings suggest that the increased Mi-2 expression during the inflammatory process of atopic dermatitis could be related to the seroconversion of anti-Mi-2 antibody in the present case. It has also been demonstrated that UV radiation increases the expression of TIF1-γ, similar to Mi-2 expression (McAvera and Crawford 2020). Taken together, it is possible that preexisting chronic dermatitis may contribute to the occurrence of both anti-Mi-2 and anti-TIF1-γ antibodies, and JDM. Although the present case experienced the relief of pruritus by topical steroid treatments, these dermatological manifestations can be associated with JDM resulting in the delay of diagnosis or treatment against JDM. The cutaneous findings of JDM may mimic atopic dermatitis (Tuen et al. 2013). Alternatively, the present case report suggests that atypical skin eruption in a child with no atopic history and no new trigger could be unusual and should not be misdiagnosed as atopic dermatitis in even the presence of eosinophilia or elevated serum levels of IgE. Further accumulation of such cases is needed to clarify the overall features of patients who have both anti-Mi-2 and anti-TIF1-γ antibodies.
In conclusion, this present case report suggests that anti-Mi-2 antibody-positive patients can be associated with classical DM without cancer even with the simultaneous presence of anti-TIF1-γ antibodies. Coexisting anti-TIF1-γ and anti-Mi-2 antibodies seen in this case may be related to the antibody-compatible clinical and cutaneous manifestations. Recognition of cutaneous findings in anti-TIF1-γ and Mi-2-positive patients may allow for a more accurate and timely diagnosis and effective treatment of patients with JDM.
Acknowledgments
The authors are grateful to Enago (https://www.enago.jp) for the English language review.
Conflict of Interest
The authors declare no conflict of interest.
References
-
Burd,
C.J.,
Kinyamu,
H.K.,
Miller,
F.W. &
Archer,
T.K.
(2008) UV radiation regulates Mi-2 through protein translation and stability. J. Biol. Chem., 283, 34976-34982.
-
Feldman,
B.M.,
Reichlin,
M.,
Laxer,
R.M.,
Targoff,
I.N.,
Stein,
L.D. &
Silverman,
E.D.
(1996) Clinical significance of specific autoantibodies in juvenile dermatomyositis. J. Rheumatol., 23, 1794-1797.
-
Fujimoto,
M.,
Hamaguchi,
Y.,
Kaji,
K.,
Matsushita,
T.,
Ichimura,
Y.,
Kodera,
M.,
Ishiguro,
N.,
Ueda-Hayakawa,
I.,
Asano,
Y.,
Ogawa,
F.,
Fujikawa,
K.,
Miyagi,
T.,
Mabuchi,
E.,
Hirose,
K.,
Akimoto,
N.,
et al.(2012) Myositis-specific anti-155/140 autoantibodies target transcription intermediary factor 1 family proteins. Arthritis Rheum., 64, 513-522.
-
Fujimoto,
M.,
Murakami,
A.,
Kurei,
S.,
Okiyama,
N.,
Kawakami,
A.,
Mishima,
M.,
Sato,
S.,
Seishima,
M.,
Suda,
T.,
Mimori,
T.,
Takehara,
K. &
Kuwana,
M.
(2016a) Enzyme-linked immunosorbent assays for detection of anti-transcriptional intermediary factor-1 gamma and anti-Mi-2 autoantibodies in dermatomyositis. J. Dermatol. Sci., 84, 272-281.
-
Fujimoto,
M.,
Watanabe,
R.,
Ishitsuka,
Y. &
Okiyama,
N.
(2016b) Recent advances in dermatomyositis-specific autoantibodies. Curr. Opin. Rheumatol., 28, 636-644.
-
Ghirardello,
A.,
Zampieri,
S.,
Iaccarino,
L.,
Tarricone,
E.,
Bendo,
R.,
Gambari,
P.F. &
Doria,
A.
(2005) Anti-Mi-2 antibodies. Autoimmunity, 38, 79-83.
-
Kaji,
K.,
Fujimoto,
M.,
Hasegawa,
M.,
Kondo,
M.,
Saito,
Y.,
Komura,
K.,
Matsushita,
T.,
Orito,
H.,
Hamaguchi,
Y.,
Yanaba,
K.,
Itoh,
M.,
Asano,
Y.,
Seishima,
M.,
Ogawa,
F.,
Sato,
S.,
et al. (2007) Identification of a novel autoantibody reactive with 155 and 140 kDa nuclear proteins in patients with dermatomyositis: an association with malignancy. Rheumatology (Oxford), 46, 25-28.
-
Kotobuki,
Y.,
Tonomura,
K. &
Fujimoto,
M.
(2021) Transcriptional intermediary factor 1 (TIF1) and anti-TIF1gamma antibody-positive dermatomyositis. Immunol. Med., 44, 23-29.
-
Liang,
L.,
Zhang,
Y.M.,
Chen,
H.,
Ye,
L.F.,
Li,
S.S.,
Lu,
X.,
Wang,
G.C. &
Peng,
Q.L.
(2020) Anti-Mi-2 antibodies characterize a distinct clinical subset of dermatomyositis with favourable prognosis. Eur. J. Dermatol., 30, 2.
-
Mammen,
A.L.,
Casciola-Rosen,
L.A.,
Hall,
J.C.,
Christopher-Stine,
L.,
Corse,
A.M. &
Rosen,
A.
(2009) Expression of the dermatomyositis autoantigen Mi-2 in regenerating muscle. Arthritis Rheum., 60, 3784-3793.
-
McAvera,
R.M. &
Crawford,
L.J.
(2020) TIF1 proteins in genome stability and cancer. Cancers (Basel), 12, 2094.
-
Morris,
P. &
Dare,
J.
(2010) Juvenile dermatomyositis as a paraneoplastic phenomenon: an update. J. Pediatr. Hematol. Oncol., 32, 189-191.
-
Ogawa-Momohara,
M.,
Muro,
Y. &
Akiyama,
M.
(2019) Anti-Mi-2 antibody titers and cutaneous manifestations in dermatomyositis. J. Cutan. Immunol. Allergy, 2, 49-52.
-
Pagnini,
I.,
Vitale,
A.,
Selmi,
C.,
Cimaz,
R. &
Cantarini,
L.
(2017) Idiopathic inflammatory myopathies: an update on classification and treatment with special focus on juvenile forms. Clin. Rev. Allergy Immunol., 52, 34-44.
-
Rider,
L.G. &
Nistala,
K.
(2016) The juvenile idiopathic inflammatory myopathies: pathogenesis, clinical and autoantibody phenotypes, and outcomes. J. Intern. Med., 280, 24-38.
-
Rider,
L.G.,
Shah,
M.,
Mamyrova,
G.,
Huber,
A.M.,
Rice,
M.M.,
Targoff, I.N. & Miller, F.W.; Childhood Myositis Heterogeneity Collaborative Study Group
(2013) The myositis autoantibody phenotypes of the juvenile idiopathic inflammatory myopathies. Medicine (Baltimore), 92, 223-243.
-
Shimizu,
K.,
Kobayashi,
T.,
Kano,
M.,
Hamaguchi,
Y.,
Takehara,
K. &
Matsushita,
T.
(2020) Anti-transcriptional intermediary factor 1-gamma antibody as a biomarker in patients with dermatomyositis. J. Dermatol., 47, 64-68.
-
Stuhlmuller,
B.,
Schneider,
U.,
Gonzalez-Gonzalez,
J.B. &
Feist,
E.
(2019) Disease specific autoantibodies in idiopathic inflammatory myopathies. Front. Neurol., 10, 438.
-
Takezaki,
D.,
Onishi,
S.,
Hamaguchi,
Y.,
Fujimoto,
M.,
Kohzan,
H. &
Hamada,
T.
(2020) Myositis-specific autoantibodies reacting to both Tif1gamma and Mi-2 in a patient with juvenile dermatomyositis. Acta Derm. Venereol., 100, adv00238.
-
Tansley,
S.L.,
McHugh,
N.J. &
Wedderburn,
L.R.
(2013) Adult and juvenile dermatomyositis: are the distinct clinical features explained by our current understanding of serological subgroups and pathogenic mechanisms? Arthritis Res. Ther., 15, 211.
-
Tuen,
V.C.,
Zingula,
S.N.,
Moir,
C.,
Reed,
A.M.,
Matsumoto,
J.M. &
Woodrum,
D.A.
(2013) MRI guided wire localization muscle biopsy in a child with juvenile dermatomyositis. Pediatr. Rheumatol. Online J., 11, 15.