Abstract
Chronic myeloid leukemia (CML) is triggered by t(9;22)(q34;q11.2) translocation, leading to the formation of the BCR-ABL1 fusion gene. Although the development of BCR-ABL1 tyrosine kinase inhibitors (TKIs) has dramatically improved the prognosis of CML, the disease could often relapse, presumably because leukemic stem cell fraction of CML (CML-LSC) may reside in specific niches, and also acquire an ability to resist the cytotoxic agents. Recently a study indicated that pharmacological inhibition of plasminogen activator inhibitor-1 (PAI-1, also known as SERPINE1) would cause detachment of CML-LSCs from their niche by inducing maturation of membrane-type matrix metalloprotease-1 (MT1-MMP), leading to increased susceptibility of CML-LSCs against TKIs. However, the direct antitumor effect of PAI-1 inhibition in CML remains unclear. Because PAI-1 mRNA expression was lower in CML cell line (K562) than bone marrow mononuclear cells derived from CML patients, we established K562 cell clones stably expressing exogenous PAI-1 (K562/PAI-1). We found that TM5614 treatment significantly suppressed cell proliferation and induced apoptosis in K562/PAI-1 cells, accompanied by increased activity of Furin protease, which is a known target of PAI-1. Besides processing mature MT1-MMP, Furin is in charge of cleaving the NOTCH receptor to form a heterodimer before exporting it to the cell surface membrane. In K562/PAI-1 cells, TM5614 treatment increased NOTCH1 intracellular domain (NICD) protein expression as well as NOTCH1 target of HEY1 mRNA levels. Finally, forced expression of either Furin or NICD in K562/PAI-1 cells significantly inhibited cell proliferation and induced apoptosis. Collectively, PAI-1 inhibition may have an antitumor effect by modulating the Furin/NICD pathway.
Introduction
BCR-ABL1 is a constitutively active, chimeric tyrosine kinase that arises from a reciprocal translocation between chromosome 9 and 22, t(9;22)(q34;q11.2), which is characteristic of Philadelphia chromosome-positive leukemia (Nowell and Hungerford 1960). Depending on the breakpoint on chromosome 22 at the BCR (breakpoint cluster) gene, three major isoforms of BCR-ABL1 can be produced, namely, the 185kDa, 210kDa, and 230kDa proteins typically found in acute lymphocytic leukemia, chronic myeloid leukemia (CML), and chronic neutrophilic leukemia, respectively (Walker et al. 1987; Pane et al. 1996; Amarante-Mendes et al. 2022). In all cases, the first exon of ABL1 (ABL proto-oncogene 1) on chromosome 9 is replaced by one of the BCR sequences.
CML is thought to be caused by stem cell transformation by the BCR-ABL1 fusion gene (Nowell and Hungerford 1960). The development of BCR-ABL1 tyrosine kinase inhibitors (TKIs), such as imatinib, has significantly improved CML prognosis (Druker et al. 2006; Hochhaus et al. 2017). Nevertheless, one of the major limitations of TKI therapy is the persistence of leukemia stem cells (LSCs) in the general patient population, which means that only a small proportion of patients can maintain treatment-free remission after stopping TKI treatment (Etienne et al. 2017). It has been hypothesized that CML-LSCs may possess the unique ability to resist cytotoxic agents in their specific niche (Bhatia et al. 2003; Huntly and Gilliland 2005; Corbin et al. 2011).
To overcome the limitation of CML-LSCs, it is expected that a novel therapeutic strategy will be developed. Several studies have recently focused on the inhibition of plasminogen activator inhibitor-1 (PAI-1, also known as SERPINE1) activity. PAI-1 is a proteolytic enzyme inhibitor found in vascular endothelial cells, the liver, platelets, and adipocytes, and is released into the bloodstream as a result of vascular endothelial damage and platelet disruption (Van De Craen et al. 2012). PAI-1 regulates the fibrinolytic response by specifically and immediately inhibiting urokinase/tissue-type plasminogen activator, which aids in the production of plasmin to dissolve formed clots (Ghosh and Vaughan 2012; Ismail et al. 2021). Additionally, PAI-1 inhibits the activity of Furin proprotein convertase, which is a family of serine proteases that are involved in the posttranslational processing and activation of a wide range of regulatory proteins (Bernot et al. 2011). PAI-1 inhibits Furin-dependent processing from pro-MT1-MMP (membrane-type I matrix metalloprotease) to MT1-MMP in hematopoietic stem cells (HSCs). Because MT1-MMP destroys pericellular matrix proteins and several adhesion molecules required for anchoring HSCs from their niche, inhibiting PAI-1 activity may promote HSC motility, resulting in HSC detachment from their niche (Yahata et al. 2017). Intriguingly, a recent study indicated that the PAI-1 inhibitor augmented motility of CML-LSCs through similar mechanisms, which may lead to increased sensitivity to the TKIs (Yahata et al. 2021). Nevertheless, the potential direct antitumor effect of PAI-1 inhibition in CML remains to be elucidated.
Hence, in the present study, we assessed the effect of the PAI-1 inhibitor (TM5614) on the basis of K562 CML cells.
Materials and Methods
Clinical sample
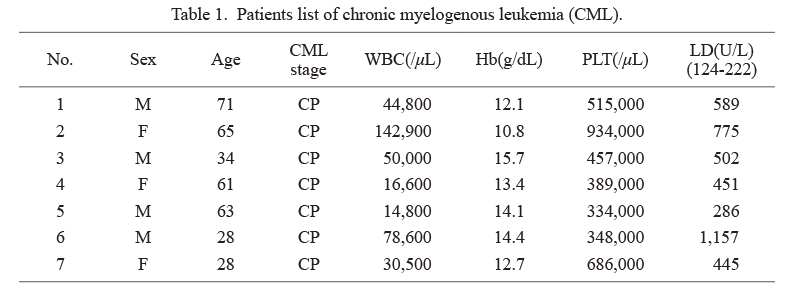
After obtaining written informed consent, bone marrow samples were obtained from seven patients (Table 1) with CML in the chronic phase. Fresh bone marrow samples were overlaid on Ficoll-Paque PLUS to obtain bone marrow mononuclear cells (GE Healthcare, Uppsala, Sweden). Samples were centrifuged at 1,500 rpm for 30 min at room temperature. After removing the upper layer, the mononuclear cells were obtained and were used for the analyses.
The genetic analysis was approved by the Ethical Committee of Tohoku University Graduate School of Medicine (Approval No. 2021-1-805), and was in accordance with the Helsinki Declaration.
Cell culture
In a humidified incubator at 37°C with 5% CO2, the K562 human CML cell line was grown in RPMI-1640 medium containing 10% fetal bovine serum (Biowest, Miami, FL, USA) and 1% penicillin-streptomycin (Sigma, St. Louis, MO, USA).
PAI-1 inhibitor
TM5614 was dissolved with dimethyl sulfoxide (Sigma), and used to inhibit PAI-1 activity (Yamaoka et al. 2018). In this study, TM5614 was treated at a concentration of 62.7 µM, which was calculated on the basis of average maximum plasma concentration (Cmax: 28 µg/mL) by repeated-dose study (120 mg/day, 7 days).
Retroviral overexpression of PAI-1
Retroviral overexpression of FOG1 and GATA-2 was conducted using pBABE-puro vector (Fujiwara et al. 2017). The retroviral vector and the env (envelope glycoprotein) gene from the vesicular stomatitis virus (VSV-G) were cotransfected into PLAT-GP Packaging Cell Lines with FuGene HD (Promega, Madison, WI, USA). Seventy-two hours after transfection, the viral supernatant was used for infection. After spin infection into K562 cells at 3,400 rpm for 2 h, the cells were cultured with the medium containing 1 µg/mL Puromycin (Sigma) for selection of the transduced cells. Single-cell clones were subsequently isolated with the limiting dilution method.
Real-time quantitative reverse transcription-polymerase chain reaction (RT-PCR)
Total RNA was purified using TRIzol (Invitrogen, Waltham, MA, USA), and 1 μg of purified total RNA was used to synthesize complementary DNA (cDNA) with ReverTra Ace qPCR RT Master Mix (TOYOBO, Osaka, Japan). Reaction mixtures (20 μL) for real-time quantitative RT-PCR comprised 2 μL of cDNA, 10 μL of Quantitect SYBR Green PCR Master Mix (QIAGEN, Venlo, Netherlands), and appropriate primers. Product accumulation was monitored by measuring SYBR Green fluorescence and normalized relative to GAPDH messenger RNA (mRNA). Table 2 shows the sequences for primers.
To calculate mRNA expression levels, an amplified cDNA fragment of each gene was cloned into the pGEM™-T Easy Vector (Promega) and was used as an internal standard in quantitative RT-PCR. The plasmid copy number was calculated as follows: copy number (copy/μL) = 6.02 × 1023 × [plasmid DNA concentration (μg/μL)] × 10−6 / [total plasmid size (base pair)] × 660, as described previously (Fujiwara et al. 2014).
Flow cytometry
Cells were analyzed using a FACSAria II flow cytometer (Becton Dickinson, Franklin Lakes, NJ, USA), and data were analyzed using FACSDiva (Becton Dickinson) or FlowJo software (TreeStar, Ashland, OR, USA).
Cells were washed with cold phosphate-buffered saline and resuspended in Annexin V Binding Buffer (eBioscience, San Diego, CA, USA) before being incubated with APC-Annexin V (eBioscience) and propidium iodide for fluorescence-activated cell sorting (FACS)-based apoptosis analysis (PI; eBioscience). Cell proliferation was measured with an APC BrdU Flow Kit (BD Pharmingen, San Jose, CA, USA), as previously described (Saito et al. 2019). Antihuman MMP-14/MT1-MMP antibody (clone 128527; R&D systems, Minneapolis, MN, USA) was used to detect cell surface MT1-MMP.
Microarray analysis
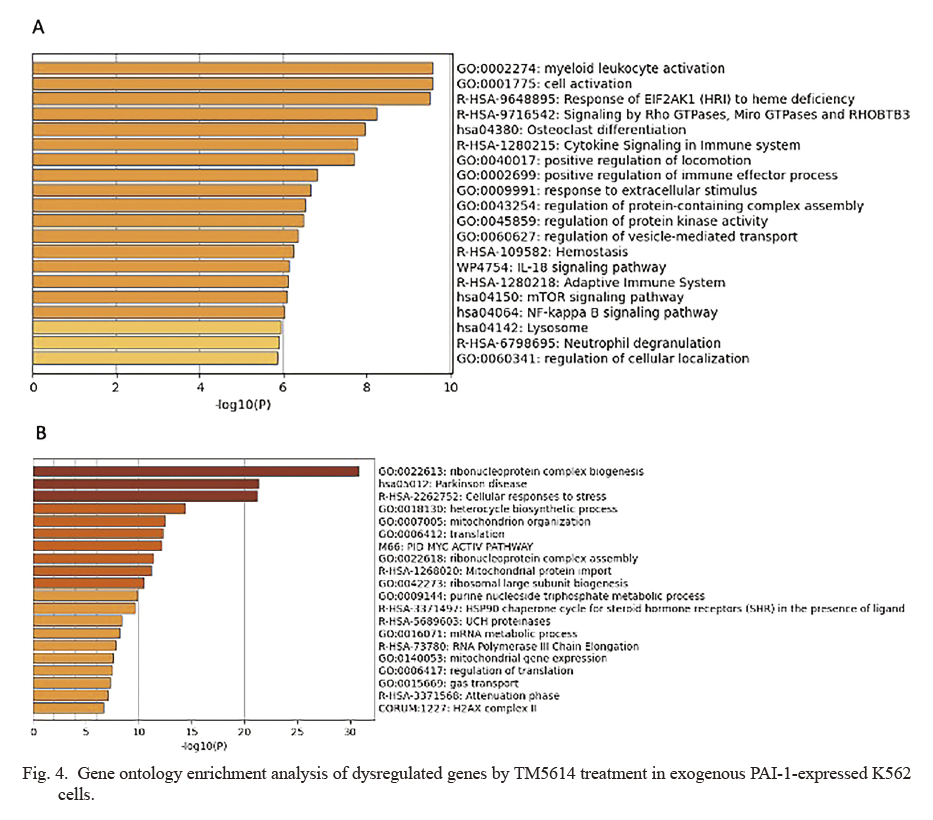
Human Oligo chip 25k (Toray, Tokyo, Japan) was used for expression profiling (Saito et al. 2019). For global normalization, the background value was subtracted and subsequently adjusted to the average signal value of 25. We selected genes showing > 100 of global normalization. Metascape (https://metascape.org) was used for GO analysis (Zhou et al. 2019). The microarray data was deposited at Tohoku University Repository (http://hdl.handle.net/10097/00134680).
Assays for PAI-1 and Furin
Cellular PAI-1 concentrations were determined using the Human PAI1 SimpleStep ELISA® Kit (SERPINE1) (Abcam, Cambridge, UK), and enzymatic activities for PAI-1 and Furin were determined using the Human PAI1 ELISA Kit and the SensoLyte® Rh110 Furin Activity Assay Kit-Fluorimatric (ANASPEC, Fremont, CA, USA), respectively.
Western blotting
Whole-cell extracts were used for Western blotting, as previously described (Fujiwara et al. 2017). The following primary antibodies were used: α-Tubulin (CP06; EMD-Millipore, Billerica, MA, USA), NOTCH1 (clone D1E11; Cell Signaling Technologies, Danvers, MA, USA), and FURIN (Abcam).
Statistical analyses
Statistical significance was assessed using a two-sided Student’s t-test. In all analyzes, p < 0.05 was considered statistically significant.
Results and Discussion
Establishment of K562 cells stably expressing exogenous PAI-1
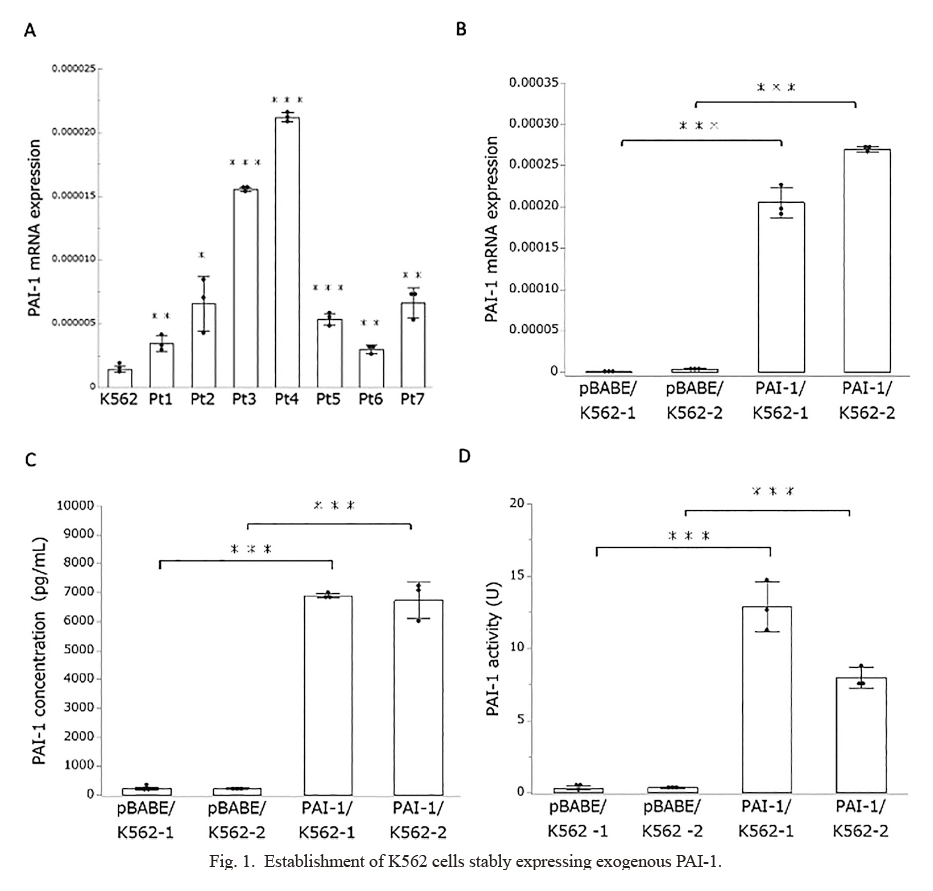
The purpose of this study was to evaluate the potential antitumor effect of PAI-1 inhibition (TM5614) in CML. We used K562 cells, a widely used cell line derived from CML patients, in this study (Collins et al. 1977). First, we used quantitative RT-PCR to assess the level of PAI-1 expression in K562 cells and primary bone marrow mononuclear cells derived from CML patients at the time of diagnosis (Table 1). PAI-1 mRNA expression levels were significantly higher in primary CML samples than in K562 cells, as shown in Fig. 1A. Although the impact of endogenous PAI-1 expression levels on CML biology is unknown, we established two independent K562 clones stably expressing exogenous PAI-1 (PAI-1/K562-1 and PAI-1/K562-2) (Fig. 1B). We further confirmed increased cellular PAI-1 protein levels (Fig. 1C) as well as PAI-1 activity (Fig. 1D) in PAI-1 expressing clones than blank vector expressed control cells (pBABE/K562-1 and pBABE-2/K562-2). In this study, we used PAI-1 overexpressed K562 cell clones as CML models for further analyses.
We assessed the effect of the PAI-1 inhibitor (TM5614) on CML cells. Cells were treated with TM5614 in PAI-1/K562-1 and PAI-1/K562-2 cells at a concentration of 62.7 μM, applying the dosages based on Cmax via repeated-dose study (data not shown). On day 3 of TM5614 treatment, we confirmed reduced PAI-1 concentration (Fig. 2A) and PAI-1 activity (Fig. 2B). As shown in Fig. 2C, inhibiting PAI-1 significantly reduces CML cell proliferation, which is accompanied by an increase in the cell cycle regulator p21 (Fig. 2D). The frequency of Annexin V-positive cells was then determined using FACS, which reflected the expression of the apoptosis marker phosphatidylserine (Segawa and Nagata 2015). The rates of early and late apoptosis processes, as measured by PI-Annexin V+ and PI+Annexin V+, respectively (Fig. 3A), were significantly increased (Fig. 3B). To estimate the molecular mechanisms of suppressed proliferation as well as increased apoptosis by PAI-1 inhibition, we conducted an expression profiling analysis on the basis of TM5614-treated PAI-1/K562-1 cells. TM5614 treatment caused a more than 1.5-fold upregulation of 129 genes and downregulation of 53 genes (http://hdl.handle.net/10097/00134680). Myeloid leukocyte activation, the NF-kappa B signaling pathway, and mitochondrion organization were all significantly enriched in the GO analysis (Fig. 4A, B). We discovered that TM5614 treatment increased the expression of PU.1 (also known as SPI1), a gene associated with myeloid leukocyte activation (http://hdl.handle.net/10097/00134680). PU.1 is known as a myeloid-lymphoid-promoting transcription factor (Scott et al. 1994; Fujiwara et al. 2017). A previous study indicated that forced PU.1 expression reduced growth rates of K562 cells (Delgado et al. 1998). While the effect of TM5614 in regulating NF-kappa B signaling remains elusive, this pathway is involved in BCR-ABL1-mediated leukemogenesis (Carrà et al. 2016), and also associated with cell death of CML cells (Lounnas et al. 2009). Additionally, we observed significant downregulation of several antiapoptotic genes associated with mitochondrion organization, including HSPA1A (HSP70) (Mosser et al. 2000), SOD1 (Zelko et al. 2002), and HSP90AA1 (Niu et al. 2021) (http://hdl.handle.net/10097/00134680), which may contribute to the increased apoptosis caused by TM5614 treatment. As described later, we focused on Furin/NOTCH signaling pathway as an important downstream effector of TM5614. However, the analysis failed to identify significant enrichment of genes involved in the Furin/NOTCH signaling pathway. It might be possible that altered myeloid gene pathway derived from the pathway analysis could reflect the activation of NOTCH signaling by TM5614. Nonetheless, to determine how PAI-1 inhibition affects CML biology, more research is needed.

PAI-1 is known as an intracellular inhibitor of Furin activity (Bernot et al. 2011). We noticed that protein levels of Furin were moderately induced by TM5614 treatment (Fig. 5A). In addition, we demonstrated that PAI-1 inhibition by TM5614 significantly augmented Furin activity in both PAI-1/K562-1 and PAI-1/K562-2 cells (Fig. 5B). The previous study demonstrated that Furin is responsible for processing from pro-MT1-MMP to MT1-MMP in HSCs (Yahata et al. 2017), and we confirmed that TM5614 treatment increased cell surface MT1-MMP expression levels (Fig. 5C).
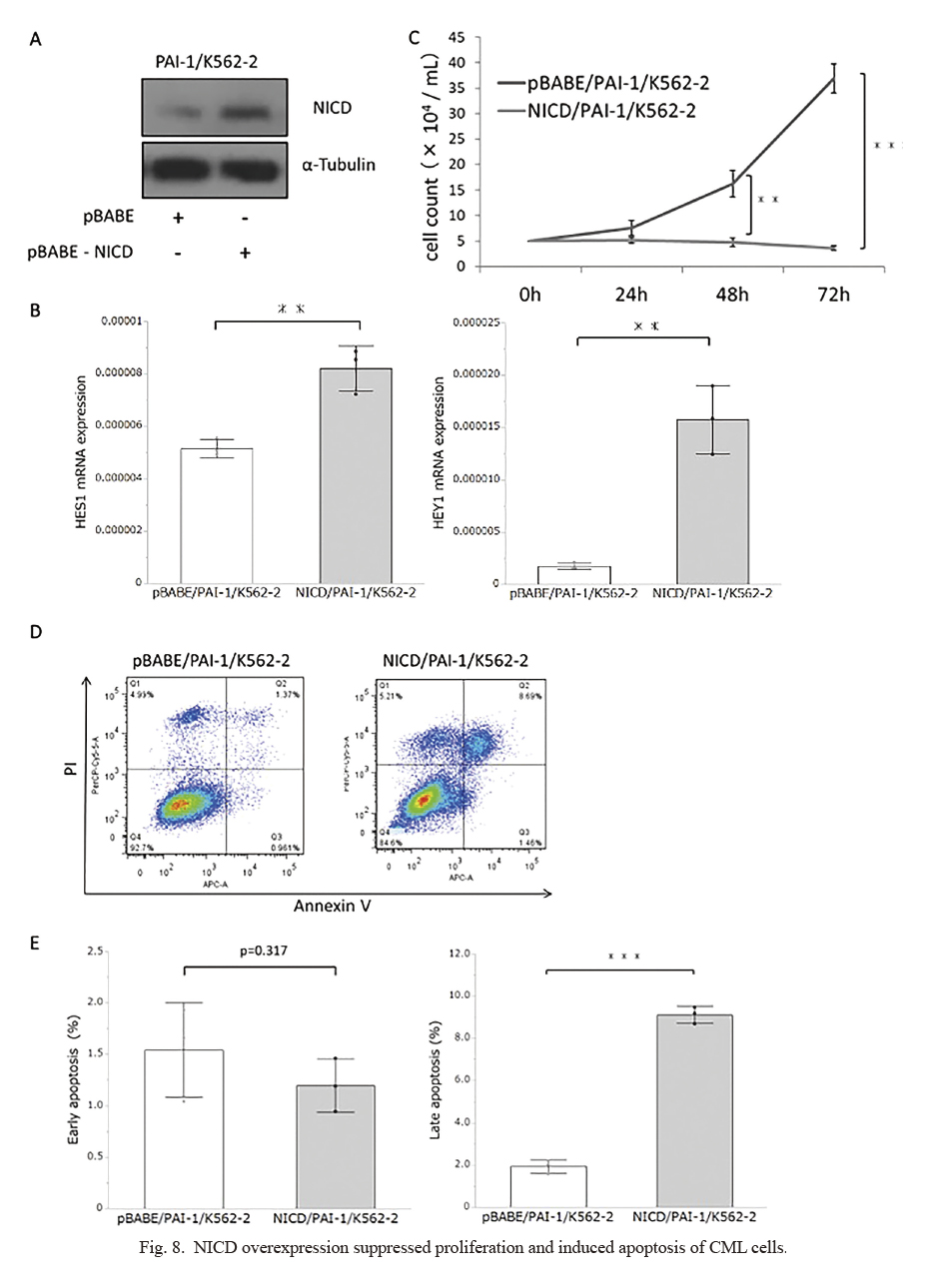
Then, we looked to see if increased Furin activity could mimic the effect of TM5614 in CML cells. We overexpressed Furin in PAI-1/K562-2 cells for this purpose (Fig. 6A) and confirmed increased Furin activity (Fig. 6B). Enhanced Furin activity significantly suppressed cell proliferation (Fig. 6C), and increased the rate of late apoptosis (Fig. 6D, E). These findings suggested that inhibiting PAI-1 increased Furin activity, suppressing proliferation and inducing apoptosis in CML cells. Besides Furin, PAI-1 could inhibit other serine proteases, such as urokinase/tissue-type plasminogen activator (Ismail et al. 2021), which are essential regulatory proteins of the fibrinolytic system. However, the impact of the altered fibrinolytic activity on CML biology has yet to be determined. It has been known that Furin is involved in NOTCH signaling, which regulates cell fate as well as other fundamental processes such as hematopoiesis (Wu and Bresnick 2007; McCarter et al. 2018). Furin’s proteolytic activity is responsible for processing the NOTCH receptor into a heterodimer, which is then exported to the cell surface membrane. When the Notch receptor interacts with its ligands on neighboring cells, it activates NOTCH signaling by releasing the NOTCH intracellular domain via γ-secretase-mediated proteolytic cleavage. The NICD enters the nucleus and activates target genes like HES1 and HEY1 (Wu and Bresnick 2007; McCarter et al. 2018). Mammalian NOTCH receptors (NOTCH1, 2, 3, and 4) make up the mammalian NOTCH system (Wu and Bresnick 2007; McCarter et al. 2018). Thus, we compared the expression levels of NOTCH receptors in CML cells. As shown in Fig. 7A, NOTCH1 was abundantly expressed in both PAI-1/K562-1 and PAI-1/K562-2 cells, whereas NOTCH3 and NOTCH4 expression levels were low to almost undetectable in these cells. Hence, we evaluated the expression level of NICD derived from NOTCH1 intracellular domain (NICD), demonstrating that TM5614 treatment resulted in increased expression levels of NICD in both PAI-1/K562-1 and PAI-1/K562-2 (Fig. 7B), and concomitantly, the expressions for HES1 and HEY1 were also significantly upregulated by the treatment (Fig. 7C). Also, when Furin was overexpressed in PAI-1/K562-2 cells (Fig. 6A), we confirmed the accumulation of NICD (Fig. 7D) and increased expression for HES1 and HEY1 (Fig. 7E). Moreover, we overexpressed NICD in PAI-1/K562-2 cells (Fig. 8A) and found increased HES1 and HEY1 expression (Fig. 8B). We found significantly suppressed cell proliferation (Fig. 8C) and significantly increased late apoptosis (Fig. 8D, E) by NICD overexpression, which was consistent with the findings based on Furin overexpression in CML cells (Fig. 6). Taken together, PAI-1 inhibition could exert an antitumor effect partly by modulating Furin/NICD pathway.
We demonstrated that TM5614 treatment in PAI-1/K562 cells significantly increased both early and late apoptotic cells (Fig. 3B), while either Furin or NICD overexpression induced only the rate of late apoptosis (Fig. 6D, E), seemingly inconsistent findings. Early apoptosis is associated with sudden removal of the survival signals or disassociation from neighboring cells, and subsequently moves into late apoptosis, when the plasma membrane becomes permeabilized (Poon et al 2010). In our analysis, Furin and NICD were stably overexpressed in the PAI-1/K562-2 cells, which might preclude the sensitive detection of early apoptotic cells. To address the issue, an establishment of inducible overexpression system of either Furin or NICD would be preferred.
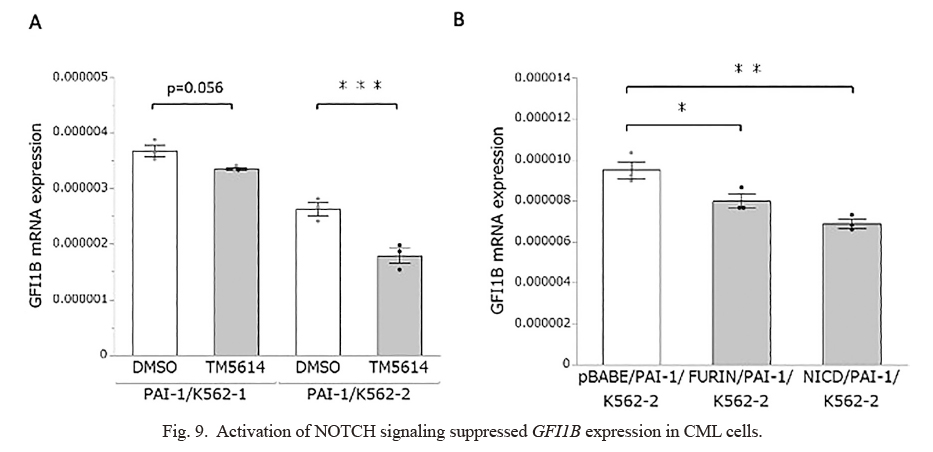
NOTCH1 is also known as a major oncogene because activating NOTCH1 mutations are found in the majority of patients with T-cell acute lymphoblastic leukemia (T-ALL) (Grabher et al. 2006). Furthermore, mice constitutively expressing NICD develop T-ALL (Thandapani et al. 2022). A previous study, conversely, found that NOTCH1 signaling could inhibit the growth of K562 cells (Yin et al. 2009), which was consistent with our findings. Nevertheless, the molecular mechanism by which NOTCH1 signaling has seemingly opposing biological effects remains unknown. Another study found that inhibiting NOTCH signaling in mouse HSCs caused an abnormal accumulation of granulocyte/monocyte progenitors as well as the induction of a chronic myelomonocytic leukemia-like phenotype (Klinakis et al. 2011), indicating that NOTCH signaling may exert tumor-suppressor function in the context of myeloid leukemia. The study further indicated that NOTCH signaling suppressed an extensive myeloid gene expression through the induction of the transcriptional repressor HES1 (Klinakis et al. 2011). Among the NOTCH1-suppressed myeloid genes, we focused on the GFI1B gene, which encodes a transcription factor responsible for the development of megakaryocytes, platelets, and erythrocytes (Beauchemin and Möröy 2020). A previous study indicated that silencing of GFI1B is associated with antileukemic effects against K562 cells as well as primary CML cells (Koldehoff et al. 2013). Intriguingly, our expression profiling analysis identified GFI1B downregulation via TM5614 treatment (http://hdl.handle.net/10097/00134680), and quantitative RT-PCR analysis confirmed the downregulation of GFI1B in TM5614-treated CML cells (Fig. 9A) as well as Furin- or NICD-overexpressed CML cells (Fig. 9B). Consequently, we hypothesize that activating NOTCH signaling has an antileukemic effect on CML cells, at least in part by suppressing key myeloid-related genes such as GFI1B. Nevertheless, additional studies, such as expression profiling with NICD-induced CML cells, would be required to reveal the detailed molecular mechanism.
There are several limitations in this study. One limitation of the study is that an in vitro experiment based on CML primary culture would be preferable for testing the effect of TM5614. We are unable to obtain additional clinical samples for the experiments due to a lack of available resources. Additionally, the potential toxicity of TM5614 against normal hematopoietic cells has not been evaluated. In this regard, PAI-1 inhibitors, including TM5614, were given to mice with no obvious hematological side effects (Ibrahim et al. 2014; Yahata et al. 2017, 2021). More importantly, it has remained unknown regarding the pathophysiological significance of PAI-1 expression in CML. We established K562 clones expressing exogenous PAI-1, because PAI-1 expression levels were lower than clinical CML samples (Fig. 1A), and PAI-1 expression level and its activity in K562 cells were around the sensitivity limit of determination in our analyses (Fig. 1C, D). However, we noticed that TM5614 treatment also significantly inhibited cell proliferation of parental K562 cells (data not shown). Thus, we speculate that inhibition of endogenous PAI-1 activity by TM5614 might suffice to exert antitumor effect, even in parental K562 cells. Nevertheless, based on our series of observations, we believe that TM5614-mediated modulation of Furin/NOTCH signaling would have an important role in exerting antitumor effect in CML.
In summary, we found that inhibiting PAI-1 had a direct antitumor effect in CML cells. Our findings would add to the evidence and rationale for PAI-1 blockade as a treatment option for CML.
Acknowledgments
We thank the members of the Biomedical Research Core of Tohoku University School of Medicine for their support.
Conflict of Interest
The authors declare no conflict of interest.
References
-
Amarante-Mendes,
G.P.,
Rana,
A.,
Datoguia,
T.S.,
Hamerschlak,
N. &
Brumatti,
G.
(2022) BCR-ABL1 tyrosine kinase complex signaling transduction: challenges to overcome resistance in chronic myeloid leukemia. Pharmaceutics, 14, 215.
-
Beauchemin,
H. &
Möröy,
T.
(2020) Multifaceted actions of GFI1 and GFI1B in hematopoietic stem cell self-renewal and lineage commitment. Front. Genet., 11, 591099.
-
Bernot,
D.,
Stalin,
J.,
Stocker,
P.,
Bonardo,
B.,
Scroyen,
I.,
Alessi,
M.C. &
Peiretti,
F.
(2011) Plasminogen activator inhibitor 1 is an intracellular inhibitor of furin proprotein convertase. J. Cell Sci., 124, 1224-1230.
-
Bhatia,
R.,
Holtz,
M.,
Niu,
N.,
Gray,
R.,
Snyder,
D.S.,
Sawyers,
C.L.,
Arber,
D.A.,
Slovak,
M.L. &
Forman,
S.J.
(2003) Persistence of malignant hematopoietic progenitors in chronic myelogenous leukemia patients in complete cytogenetic remission following imatinib mesylate treatment. Blood, 101, 4701-4707.
-
Carrà,
G.,
Torti,
D.,
Crivellaro,
S.,
Panuzzo,
C.,
Taulli,
R.,
Cilloni,
D.,
Guerrasio,
A.,
Saglio,
G. &
Morotti,
A.
(2016) The BCR-ABL/NF-kappaB signal transduction network: a long lasting relationship in Philadelphia positive Leukemias. Oncotarget, 7, 66287-66298.
-
Collins,
S.J.,
Gallo,
R.C. &
Gallagher,
R.E.
(1977) Continuous growth and differentiation of human myeloid leukaemic cells in suspension culture. Nature, 270, 347-349.
-
Corbin,
A.S.,
Agarwal,
A.,
Loriaux,
M.,
Cortes,
J.,
Deininger,
M.W. &
Druker,
B.J.
(2011) Human chronic myeloid leukemia stem cells are insensitive to imatinib despite inhibition of BCR-ABL activity. J. Clin. Invest., 121, 396-409.
-
Delgado,
M.D.,
Gutierrez,
P.,
Richard,
C.,
Cuadrado,
M.A.,
Moreau-Gachelin,
F. &
Leon,
J.
(1998) Spi-1/PU.1 proto-oncogene induces opposite effects on monocytic and erythroid differentiation of K562 cells. Biochem. Biophys. Res. Commun., 252, 383-391.
-
Druker,
B.J.,
Guilhot,
F.,
O’Brien,
S.G.,
Gathmann,
I.,
Kantarjian,
H.,
Gattermann,
N.,
Deininger,
M.W.,
Silver,
R.T.,
Goldman,
J.M.,
Stone,
R.M.,
Cervantes,
F.,
Hochhaus,
A.,
Powell,
B.L.,
Gabrilove,
J.L.,
Rousselot,
P.,
et al.(2006) Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N. Engl. J. Med., 355, 2408-2417.
-
Etienne,
G.,
Guilhot,
J.,
Rea,
D.,
Rigal-Huguet,
F.,
Nicolini,
F.,
Charbonnier,
A.,
Guerci-Bresler,
A.,
Legros,
L.,
Varet,
B.,
Gardembas,
M.,
Dubruille,
V.,
Tulliez,
M.,
Noel,
M.P.,
Ianotto,
J.C.,
Villemagne,
B.,
et al.(2017) Long-term follow-up of the French stop imatinib (STIM1) study in patients with chronic myeloid leukemia. J. Clin. Oncol., 35, 298-305.
-
Fujiwara,
T.,
Okamoto,
K.,
Niikuni,
R.,
Takahashi,
K.,
Okitsu,
Y.,
Fukuhara,
N.,
Onishi,
Y.,
Ishizawa,
K.,
Ichinohasama,
R.,
Nakamura,
Y.,
Nakajima,
M.,
Tanaka,
T. &
Harigae,
H.
(2014) Effect of 5-aminolevulinic acid on erythropoiesis: a preclinical in vitro characterization for the treatment of congenital sideroblastic anemia. Biochem. Biophys. Res. Commun., 454, 102-108.
-
Fujiwara,
T.,
Sasaki,
K.,
Saito,
K.,
Hatta,
S.,
Ichikawa,
S.,
Kobayashi,
M.,
Okitsu,
Y.,
Fukuhara,
N.,
Onishi,
Y. &
Harigae,
H.
(2017) Forced FOG1 expression in erythroleukemia cells: induction of erythroid genes and repression of myelo-lymphoid transcription factor PU.1. Biochem. Biophys. Res. Commun., 485, 380-387.
-
Ghosh,
A.K. &
Vaughan,
D.E.
(2012) PAI-1 in tissue fibrosis. J. Cell. Physiol., 227, 493-507.
-
Grabher,
C.,
von Boehmer,
H. &
Look,
A.T.
(2006) Notch 1 activation in the molecular pathogenesis of T-cell acute lymphoblastic leukaemia. Nat. Rev. Cancer, 6, 347-359.
-
Hochhaus,
A.,
Larson,
R.A.,
Guilhot,
F.,
Radich,
J.P.,
Branford,
S.,
Hughes,
T.P.,
Baccarani,
M.,
Deininger,
M.W.,
Cervantes,
F.,
Fujihara,
S.,
Ortmann,
C.E.,
Menssen,
H.D.,
Kantarjian,
H.,
O'Brien,
S.G.,
Druker,
B.J.,
et al.(2017) Long-term outcomes of imatinib treatment for chronic myeloid leukemia. N. Engl. J. Med., 376, 917-927.
-
Huntly,
B.J. &
Gilliland,
D.G.
(2005) Leukaemia stem cells and the evolution of cancer-stem-cell research. Nat. Rev. Cancer, 5, 311-321.
-
Ibrahim,
A.A.,
Yahata,
T.,
Onizuka,
M.,
Dan,
T.,
Van Ypersele De Strihou,
C.,
Miyata,
T. &
Ando,
K.
(2014) Inhibition of plasminogen activator inhibitor type-1 activity enhances rapid and sustainable hematopoietic regeneration. Stem Cells, 32, 946-958.
-
Ismail,
A.A.,
Shaker,
B.T. &
Bajou,
K.
(2021) The plasminogen-activator plasmin system in physiological and pathophysiological angiogenesis. Int. J. Mol. Sci., 23, 337.
-
Klinakis,
A.,
Lobry,
C.,
Abdel-Wahab,
O.,
Oh,
P.,
Haeno,
H.,
Buonamici,
S.,
van De Walle,
I.,
Cathelin,
S.,
Trimarchi,
T.,
Araldi,
E.,
Liu,
C.,
Ibrahim,
S.,
Beran,
M.,
Zavadil,
J.,
Efstratiadis,
A.,
et al.
(2011) A novel tumour-suppressor function for the Notch pathway in myeloid leukaemia. Nature, 473, 230-233.
-
Koldehoff,
M.,
Zakrzewski,
J.L.,
Beelen,
D.W. &
Elmaagacli,
A.H.
(2013) Additive antileukemia effects by GFI1B- and BCR-ABL-specific siRNA in advanced phase chronic myeloid leukemic cells. Cancer Gene Ther., 20, 421-427.
-
Lounnas,
N.,
Frelin,
C.,
Gonthier,
N.,
Colosetti,
P.,
Sirvent,
A.,
Cassuto,
J.P.,
Berthier,
F.,
Sirvent,
N.,
Rousselot,
P.,
Dreano,
M.,
Peyron,
J.F. &
Imbert,
V.
(2009) NF-kappaB inhibition triggers death of imatinib-sensitive and imatinib-resistant chronic myeloid leukemia cells including T315I Bcr-Abl mutants. Int. J. Cancer, 125, 308-317.
-
McCarter,
A.C.,
Wang,
Q. &
Chiang,
M.
(2018) Notch in leukemia. Adv. Exp. Med. Biol., 1066, 355-394.
-
Mosser,
D.D.,
Caron,
A.W.,
Bourget,
L.,
Meriin,
A.B.,
Sherman,
M.Y.,
Morimoto,
R.I. &
Massie,
B.
(2000) The chaperone function of hsp70 is required for protection against stress-induced apoptosis. Mol. Cell. Biol., 20, 7146-7159.
-
Niu,
M.,
Zhang,
B.,
Li,
L.,
Su,
Z.,
Pu,
W.,
Zhao,
C.,
Wei,
L.,
Lian,
P.,
Lu,
R.,
Wang,
R.,
Wazir,
J.,
Gao,
Q.,
Song,
S. &
Wang,
H.
(2021) Targeting HSP90 inhibits proliferation and induces apoptosis through AKT1/ERK pathway in lung cancer. Front. Pharmacol., 12, 724192.
-
Nowell,
P.C. &
Hungerford,
D.A.
(1960) Chromosome studies on normal and leukemic human leukocytes. J. Natl. Cancer Inst., 25, 85-109.
-
Pane,
F.,
Frigeri,
F.,
Sindona,
M.,
Luciano,
L.,
Ferrara,
F.,
Cimino,
R.,
Meloni,
G.,
Saglio,
G.,
Salvatore,
F. &
Rotoli,
B.
(1996) Neutrophilic-chronic myeloid leukemia: a distinct disease with a specific molecular marker (BCR/ABL with C3/A2 junction). Blood, 88, 2410-2414.
-
Poon,
I.K.,
Hulett,
M.D. &
Parish,
C.R.
(2010) Molecular mechanisms of late apoptotic/necrotic cell clearance. Cell Death Differ., 17, 381-397.
-
Saito,
K.,
Fujiwara,
T.,
Hatta,
S.,
Morita,
M.,
Ono,
K.,
Suzuki,
C.,
Fukuhara,
N.,
Onishi,
Y.,
Nakamura,
Y.,
Kawamata,
S.,
Shimizu,
R.,
Yamamoto,
M. &
Harigae,
H.
(2019) Generation and molecular characterization of human ring sideroblasts: a key role of ferrous iron in terminal erythroid differentiation and ring sideroblast formation. Mol. Cell. Biol., 39, e00387-18.
-
Scott,
E.W.,
Simon,
M.C.,
Anastasi,
J. &
Singh,
H.
(1994) Requirement of transcription factor PU.1 in the development of multiple hematopoietic lineages. Science, 265, 1573-1577.
-
Segawa,
K. &
Nagata,
S.
(2015) An apoptotic 'eat me' signal: phosphatidylserine exposure. Trends Cell Biol., 25, 639-650.
-
Thandapani,
P.,
Kloetgen,
A.,
Witkowski,
M.T.,
Glytsou,
C.,
Lee,
A.K.,
Wang,
E.,
Wang,
J.,
LeBoeuf,
S.E.,
Avrampou,
K.,
Papagiannakopoulos,
T.,
Tsirigos,
A. &
Aifantis,
I.
(2022) Valine tRNA levels and availability regulate complex I assembly in leukaemia. Nature, 601, 428-433.
-
Van De Craen,
B.,
Declerck,
P.J. &
Gils,
A.
(2012) The biochemistry, physiology and pathological roles of PAI-1 and the requirements for PAI-1 inhibition in vivo. Thromb. Res., 130, 576-585.
-
Walker,
L.C.,
Ganesan,
T.S.,
Dhut,
S.,
Gibbons,
B.,
Lister,
T.A.,
Rothbard,
J. &
Young,
B.D.
(1987) Novel chimaeric protein expressed in Philadelphia positive acute lymphoblastic leukaemia. Nature, 329, 851-853.
-
Wu,
J. &
Bresnick,
E.H.
(2007) Bare rudiments of notch signaling: how receptor levels are regulated. Trends Biochem. Sci., 32, 477-485.
-
Yahata,
T.,
Ibrahim,
A.A.,
Hirano,
K.I.,
Muguruma,
Y.,
Naka,
K.,
Hozumi,
K.,
Vaughan,
D.E.,
Miyata,
T. &
Ando,
K.
(2021) Targeting of plasminogen activator inhibitor-1 activity promotes elimination of chronic myeloid leukemia stem cells. Haematologica, 106, 483-494.
-
Yahata,
T.,
Ibrahim,
A.A.,
Muguruma,
Y.,
Eren,
M.,
Shaffer,
A.M.,
Watanabe,
N.,
Kaneko,
S.,
Nakabayashi,
T.,
Dan,
T.,
Hirayama,
N.,
Vaughan,
D.E.,
Miyata,
T. &
Ando,
K.
(2017) TGF-beta-induced intracellular PAI-1 is responsible for retaining hematopoietic stem cells in the niche. Blood, 130, 2283-2294.
-
Yamaoka,
N.,
Murano,
K.,
Kodama,
H.,
Maeda,
A.,
Dan,
T.,
Nakabayashi,
T.,
Miyata,
T. &
Meguro,
K.
(2018) Identification of novel plasminogen activator inhibitor-1 inhibitors with improved oral bioavailability: structure optimization of N-acylanthranilic acid derivatives. Bioorg. Med. Chem. Lett., 28, 809-813.
-
Yin,
D.D.,
Fan,
F.Y.,
Hu,
X.B.,
Hou,
L.H.,
Zhang,
X.P.,
Liu,
L.,
Liang,
Y.M. &
Han,
H.
(2009) Notch signaling inhibits the growth of the human chronic myeloid leukemia cell line K562. Leuk. Res., 33, 109-114.
-
Zelko,
I.N.,
Mariani,
T.J. &
Folz,
R.J.
(2002) Superoxide dismutase multigene family: a comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures, evolution, and expression. Free Radic. Biol. Med., 33, 337-349.
-
Zhou,
Y.,
Zhou,
B.,
Pache,
L.,
Chang,
M.,
Khodabakhshi,
A.H.,
Tanaseichuk,
O.,
Benner,
C. &
Chanda,
S.K.
(2019) Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun., 10, 1523.