Regular Contribution
Rhamnazin Inhibits Hepatocellular Carcinoma Cell Aggressiveness in Vitro via Glutathione Peroxidase 4-Dependent Ferroptosis
2022 Volume 258 Issue 2 Pages 111-120

Details
2022 Volume 258 Issue 2 Pages 111-120
Ferroptosis, a newly recognized type of programmed cell death, is characterized by lipid peroxidation and implicated in multiple pathophysiological processes. Ferroptosis agonists are attracting tremendous attention for the clinical management of malignancy. We uncovered that rhamnazin exerted its anti-cancer property via reducing cell proliferation and invasion in hepatocellular carcinoma (HCC) cells. The ferroptosis inhibitor ferrostatin-1 (Fer-1) partially reversed rhamnazin-triggered cell proliferation inhibition, indicating that ferroptosis contributed to the inhibitory potency of rhamnazin. Further characterization corroborated that exposure with rhamnazin, reactive oxygen species (ROS) accumulation and lipid peroxidation, and iron content were elevated in HCC cells. Mechanistically, we demonstrated that glutathione peroxidase 4 (GPX4) was involved in rhamnazin-initiated ferroptotic cell death. Overexpression of GPX4 weakened HCC cell ferroptosis caused by rhamnazin. Collectively, these results strongly suggest that rhamnazin exerts a ferroptosis-inducing role in HCC cells by inhibiting GPX4 expression.
Liver cancer is a common aggressive neoplasm with a high occurrence and high mortality, among which hepatocellular carcinoma (HCC) represents the most frequent form of liver cancer (Siegel et al. 2020). Currently, surgical resection is the predominant therapeutic approach for human liver cancer, and sorafenib, a multi-kinase inhibitor, is the only first-line anti-cancer drug (Regmi et al. 2021). Substantial evidence has established a fundamental role of ferroptosis, a novel form of regulated cell death (RCD), in several physiological processes including HCC (Zhang et al. 2019). Ferroptosis is triggered by the inactivation of cystine/glutamate transporter, solute carrier family 7 member 11 (SLC7A11)/system xc-, accumulating lipid reactive oxygen species (ROS) and the iron level, and downregulating glutathione peroxidase 4 (GPX4) (Yang et al. 2020).
Sorafenib is a potent inducer of ferroptosis rather than apoptosis, in HCC cells (Louandre et al. 2013; Galmiche et al. 2014). A combination of sorafenib and other anti-cancer drugs, especially drugs targeting molecules involved in sorafenib resistance, appears increasingly promising to overcome sorafenib resistance (Won et al. 2017; Kim et al. 2018; Li et al. 2021a). Artesunate synergizes with the pro-oxidative effects of sorafenib through facilitating lipid peroxidation, lysosomal cathepsin B/L activation, and consequent ferroptosis (Li et al. 2021b). Rhamnazin (C17H14O7), one of the polyphenolic compounds, is often extracted from Rhamnineae, Ginkgo biloba, Salix, Sea buckthorn and other medicinal plants (Yu et al. 2018). Recently, rhamnazin has been demonstrated to potentiate the chemo-therapeutic effect of sorafenib via upregulation of the p38 mitogen-activated protein kinase (p38MAPK)/caspase-3 axis and downregulating vascular endothelial growth factor receptor 2 (VEGFR2) in HCC (Habiba et al. 2022). Rhamnazin directly inhibits breast cancer cells proliferation via interruption of vascular endothelial growth factor (VEGF)-induced phosphorylation of VEGFR2 (Yu et al. 2015). Rhamnazin also inhibits proliferation and induces apoptosis of Jurkat leukemia cells in vitro (Philchenkov and Zavelevych 2015). To date, only a few investigations have been conducted on the role of rhamnazin in tumor cell apoptosis during the last decade. Given the vital role of sorafenib in ferroptosis, we firstly wished to elucidate the biological role of rhamnazin in ferroptosis.
Phenotypic experiments revealed that rhamnazin inhibited the proliferation and invasive-trait of HCC cells. Subsequently, we demonstrated that ferroptosis contributed to the inhibition effect of rhamnazin. Finally, we showed that rhamnazin exerts anti-carcinoma properties via evoking ferroptosis, and GPX4 was a crucial mediator of rhamnazin-induced ferroptosis in HCC cells.
HCC Huh7 and SMMC-7721 cell lines were confirmed by short tandem repeat (STR) testing and purchased from Cobioer Biosciences (Cobioer, Nanjing, China). Huh7 and SMMC-7721 cell lines were stored in our laboratory and the well-characterized cell lines for analyzing ferroptosis-induced HCC cell death (Bai et al. 2019; Li et al. 2021a, 2022). SMMC-7721 cell was maintained in RPMI-1640 medium (Invitrogen, Carlsbad, CA, USA) with 10% fetal bovine serum (FBS), and Huh7 cell was maintained in DMEM (Invitrogen) + 10% FBS in 5% CO2 at 37°C. Rhamnazin (> 99%) (#93815) was purchased from Sigma-Aldrich (Shanghai, China). Z-VAD-FMK (#S7023), chloroquine (#S6999), necrostatin-1 (#S8037), and ferrostatin-1 (#S7243) were purchased from Selleck (Shanghai, China). The pcDNA3.1 empty vector and GPX4 overexpression vector (pcDNA3.1-GPX4) were provided by Genepharma (Shanghai, China). Huh7 or SMMC-7721 cells (1 × 106 cells/mL) were cultured in 6-well plates overnight. After reaching 70% confluence, the indicated vectors were transfected into cells using a Lipofectamine 2000 Transfection Reagent (Invitrogen) for 48 h.
Cell proliferationCell viability was evaluated using a cell counting kit-8 (CCK-8) kit (Beyotime, Nantong, China). Cells (1 × 106 cells/mL) were seeded in 96-well plates and treated with rhamnazin. The cells in the control group were treated with dimethylsulfoxide (DMSO) only and the final DMSO concentration was 0.1%. Cells in culture medium only served as a blank group. On day 3, 100 μl of CCK-8 solution was put into each well. After 4 h, the absorbance (450 nm) was measured using a microplate reader (Thermo Fisher Scientific, Waltham, MA, USA).
Transwell assayCells (1 × 105/ml) were suspended in the medium containing different concentrations of rhamnazin and added to the upper compartment of a 24-well Transwell plate (Corning, NY, USA). Cells treated with 0.1% DMSO served as the vehicle control group. 600 μL of medium supplemented with 10% FBS was put in the lower compartment. 24 h post-treatment, the cells on the underside of the membrane were dyed with 1% crystal violet (Sigma-Aldrich, St. Louis, MO, USA) and counted on an inverted microscope. Invasiveness was determined by counting cells in five randomly selected visual fields.
Clonogenic assayBriefly, cells (1 × 103/well) were seeded in 6-well plates. The complete medium containing rhamnazin was changed every three days. Cells treated with 0.1% DMSO served as the vehicle control group. Two weeks post-treatment, the colonies were fixed and counted (Chen et al. 2020b).
Analysis of lipid peroxidation (MDA), iron, and reactive oxygen species (ROS) levelsLipid Peroxidation (MDA) was determined using an MDA assay kit (#ab118970; Abcam, CA, USA). Iron was measured by utilizing an Iron assay kit (#ab83366; Abcam). ROS production was determined using an enzyme-linked immunosorbent assay (ELISA) kit (Vazyme Biotech, Nanjing, China). Cells treated with 0.1% DMSO served as the vehicle control group. Cells in culture medium only served as a blank group. All detection processes were conducted following the kit directions.
Apoptosis assayFlow cytometry was used to detect cell apoptosis. SMMC-7721 or Huh-7 cells were seeded in a 6-well plate and cultured for 24 h with 10 μM rhamnazin or the vehicle control (“Control”, 0.1% of DMSO). The cells were treated with trypsin, washed twice with phosphate-buffered saline (PBS), and then resuspended in 200 μL binding buffer and 5 μL Annexin V-FITC for 15 min. Afterward, 10 μL propidium iodide (PI) solution and 300 μL binding buffer were added to each sample. The apoptotic cells were evaluated by flow cytometer (Accuri C6, BD Biosciences, San José, CA, USA).
Determination of GSH (reduced glutathione)/GSSG (oxidized glutathione) ratioSMMC-7721 or Huh-7 cells (2 × 105 cells/well) were seeded into 6-well plates. After treatment with rhamnazin (5, 10, and 15 µM) or the vehicle control (“Control”, 0.1% of DMSO) for 24 h at 37°C, cells were lysed. The GSH/GSSG ratio was detected using a GSH/GSSG Ratio Detection Assay kit (#10056; Beyotime), following the manufacturer's protocols (Qin et al. 2020).
MMP-2/9 activities assayThe activities of MMP-2/9 in HCC cells were analyzed using the commercially available quantitative test ELISA (#H146-1, H146-4, Nanjing Jiancheng Corp., Nanjing, China) according to the manufacturer's protocol (Wang et al. 2016).
ImmunoblottingProteins were extracted from indicated cells and subjected to electrophoresis on 8% SDS-PAGE and immediately transferred to PVDF membranes (Millipore, Burlington, MA, USA). After being blocked with 5% nonfat milk, the membranes were incubated with GPX4 (#K003083P, 1:1,000, Solarbio, Beijing, China), system xc- (#A2413,1:1,000, ABclonal, Wuhan, China) or GAPDH antibody (#K200057M, 1:1,000, Solarbio) at 4°C overnight. In the following day, the membranes were incubated with horseradish peroxidase (HRP) conjugated goat anti-rabbit IgG (#AS014, 1:10,000, Solarbio) for 2 h. The target blots were visualized using an enhanced chemiluminescence (ECL) kit (Millipore). The grey value of the protein bands was analyzed by ImageJ software (ImageJ Software Inc., Bethesda, MD, USA).
Statistical analysesStatistical analysis was performed using GraphPad Prism 7.0 (La Jolla, CA, USA) and data are shown as Mean ± SD. The statistical significance was determined using Student’s t-test. p < 0.05 was statistically significant.
To evaluate the inhibitory effects of rhamnazin in HCC cells proliferation, the SMMC-7721, and Huh-7 cells were treated with rhamnazin. CCK-8 assays showed that rhamnazin sharply inhibited the growth of HCC cells. The dose-response curves were generated to calculate the inhibitory concentrations (IC50). Rhamnazin had the potential to inhibit the growth of SMMC-7721 and Huh-7 cells with IC50 values of 15 μM and 19.8 μM, respectively (Fig. 1A, B). The obtained IC50 values for two cell lines are close to 15 μM which avoids the obvious cytotoxicity induced by rhamnazin (Wawruszak et al. 2015; Krishnan et al. 2017). Rhamnazin at 5 μM (1/3 IC50), 10 μM (2/3 IC50), and 15 μM (IC50) were used for the subsequent experiments. In the colony formation assay, rhamnazin treatment dramatically compromised the clonogenic capacity of HCC cells (Fig. 1C, D).

Rhamnazin inhibits the proliferation and colony formation in HCC cells.
(A, B) Cell viability of SMMC-7721 and Huh-7 was measured using the CCK-8 assay. (C) Rhamnazin inhibited the clone formation ability of SMMC-7721 and Huh-7 cells. (D) The quantitative analyses of colony numbers are shown. *p < 0.05, **p < 0.01 compared with control.
Subsequently, Transwell assay was applied to evaluate the suppressive effect of rhamnazin on HCC cells invasion. As presented in Fig. 2A, B, rhamnazin evidently attenuated Huh-7 and SMMC-7721 cell invasion after exposure for 24 h. Metastatic cancer cells are commonly characterized by the increased production of matrix metalloproteinase (MMP) (Rani et al. 2021). Previous studies have demonstrated that progression, invasiveness, and metastatic ability of cancer cells are correlated with MMP activity (Liu et al. 2020). Inhibition of MMP activity may provide an approach to combat cancer metastasis (Shay et al. 2015). We next detected the activities of MMP-2 and MMP-9 in HCC cells. As expected, MMP-2/9 activity was markedly suppressed by rhamnazin treatment (Fig. 2C, D). These results, taken together, exhibited that rhamnazin potently lessened HCC cells invasion in vitro.

Rhamnazin inhibits HCC cells invasion.
(A) The Transwell invasion assay showed that rhamnazin suppressed the invasion of SMMC-7721 and Huh-7 cells. Scar bar = 100 μm. (B) Quantification of the invasiveness of SMMC-7721 and Huh-7 is shown. (C, D) MMP-2 and MMP-9 in the culture supernatants were measured by ELISA. *p < 0.05, **p < 0.01 compared with control.
To elaborate on the reasons for rhamnazin-induced growth inhibition, HCC cells were exposed to rhamnazin and cell death inhibitors. Chloroquine (CQ, a potent inhibitor of autophagy), necrostatin-1 (NEC-1, a potent inhibitor of necroptosis), or Z-VAD-FMK (a pancaspase inhibitor) did not reverse the repression effect of rhamnazin in HCC cells proliferation (Fig. 3A). However, the proliferation of HCC cells was blocked by the ferroptosis inhibitor, ferrostatin-1 (Fer-1) in the presence of rhamnazin (Fig. 3A). Notably, we did not detect a significant effect on induction of HCC cell apoptosis upon treatment with rhamnazin (Fig. 3B). Ferroptosis is the process of iron (Fe2+)-dependent programmed cell death, characterized by Fe2+-dependent lipid peroxidation, and the accumulation of intracellular ROS (Yang et al. 2020). Increased intracellular Fe2+ accumulation, ROS accumulation and lipid peroxidation, such as MDA are the key features of ferroptosis (Skouta et al. 2014; Yang and Stockwell 2016). We quantified the contents of lipid ROS production, Fe2+, and MDA in Huh-7 and SMC-7721 cells after being treated with rhamnazin. Similar to erastin and sorafenib, the ROS production (Fig. 3C), MDA levels (Fig. 3D), and Fe2+ content (Fig. 3E) were elevated upon stimulation with rhamnazin in HCC cells (Dixon et al. 2012; Sun et al. 2016). Collectively, these data implicate that rhamnazin induces ferroptosis in HCC cells.
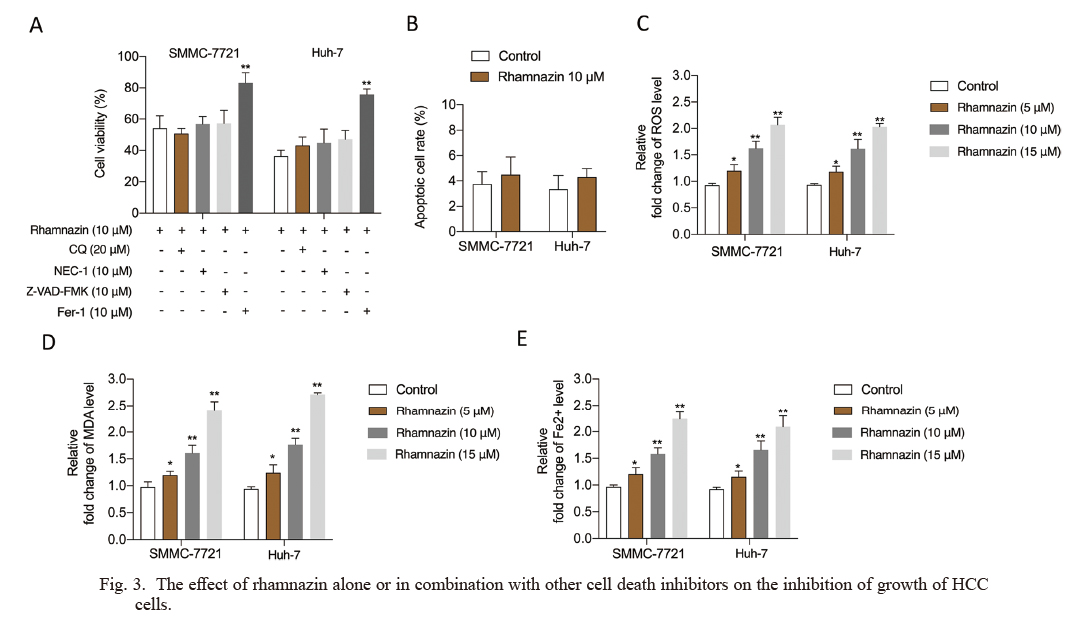
The effect of rhamnazin alone or in combination with other cell death inhibitors on the inhibition of growth of HCC cells.
(A) SMMC-7721 and Huh-7 cells were treated with rhamnazin with or without chloroquine (CQ, a potent inhibitor of autophagy), necrostatin-1 (NEC-1, a potent inhibitor of necroptosis), Z-VAD-FMK (a pancaspase inhibitor) or ferrostatin-1 (Fer-1, the ferroptosis inhibitor), and the cell viability was assayed. (B) SMMC-7721 and Huh-7 cells were treated with 10 μM rhamnazin or the vehicle control for 24 h. The apoptosis of SMMC-7721 or Huh-7 cells were determined by flow cytometry. (C-E) The ROS production, MDA level, and Fe2+ content in the culture supernatants from SMMC-7721 and Huh-7 cells were measured by ELISA. *p < 0.05, **p < 0.01 compared with control.
Ferroptosis is initiated through two major pathways: the intrinsic or enzyme-regulated pathway and the extrinsic or transporter-dependent pathway (Chen et al. 2021). The influence of rhamnazin on system xc- (a cystine/glutamic acid reverse exchange transporter) was then investigated. In the presence of rhamnazin, no significant inhibition in system xc- expression was observed (Fig. 4A), indicating that the induction of ferroptosis was, therefore, not dependent on the extrinsic pathway. In the intrinsic pathway, glutathione (GSH) is synthesized from cysteine, glutamate, and glycine (Fig. 4B). Mechanistically, cell death from ferroptosis associates with notable decreases in the cellular ratio of GSH/oxidized glutathione (GSSG). GSH and GSSG are important cellular antioxidant systems that provide an inhibitory environment for the reduction of oxidized substances (Du et al. 2021). Firstly, we clarified that rhamnazin decreased the GSH/GSSG ratio in HCC cells in a dose-dependent manner (Fig. 4C). GSH is the major antioxidant in cells and reduced GSH is utilized by glutathione peroxidase 4 (GPX4) to reduce lipid hydroperoxides and therefore protects cells against ferroptosis. To further elucidate the potential target where rhamnazin influences GSH, we measured glutamate, glycine, and cysteine levels after treatment of rhamnazin in SMMC-7721 and Huh-7 cells. The contents of these critical intermediate products were not reduced significantly by rhamnazin (Fig. 4D-F), suggesting that the target of rhamnazin lie downstream of GSH. The inactivation of GPX4 leads to accumulation of lipid peroxides and induces ferroptosis (Chen et al. 2021). We found that the protein level of GPX4 was decreased by rhamnazin (Fig. 4G, H), which suggests that GPX4 is a potential target of rhamnazin.

Effects of rhamnazin on the expression of GPX4.
(A) Protein levels of system xc- were analyzed by immunoblotting in SMMC-7721 and Huh-7 cells with or without rhamnazin treatment. (B) The ferroptosis-related metabolic axis from cystine to GSH. (C) SMMC-7721 and Huh-7 cells were treated with rhamnazin or the vehicle control for 24 h. The relative abundance of GSH/GSSG level was tested. *p < 0.05, **p < 0.01 compared with control. (D-F) The levels of glycine, glutamate, and cysteine were measured in SMMC-7721 and Huh-7 cells with or without rhamnazin treatment. (G) Protein levels of GPX4 were analyzed by immunoblotting in SMMC-7721 and Huh-7 cells with or without rhamnazin treatment. (H) The protein bands were quantified by gray scanning. *p < 0.05, **p < 0.01 compared with 0 μM rhamnazin.
Our results indicated that rhamnazin decreased GPX4 expression in HCC cells. Here, we further examined whether GPX4 was involved in the effects of rhamnazin-induced ferroptosis. GPX4 was upregulated by transfections with pcDNA3.1-GPX4 in SMMC-7721 and Huh-7 followed by rhamnazin treatment (Fig. 5A). The inhibiting effects of rhamnazin on HCC cells were effectively blocked by GPX4 overexpression, as demonstrated by cell proliferation (Fig. 5B) and colony formation (Fig. 5C). We, therefore, tested whether GPX4 overexpression could reverse ferroptotic phenotype caused by rhamnazin. Consistently, the amount of ROS (Fig. 5D), MDA level (Fig. 5E), and Fe2+ content (Fig. 5F) increased by rhamnazin were reversed upon GPX4 overexpression. All these observations implied the involvement of GPX4 in the effects of rhamnazin on HCC cells ferroptosis.

Ferroptosis contributes to rhamnazin-induced growth inhibition in HCC cells.
(A) The expression of GPX4 in SMMC-7721 and Huh-7 cells transfected with pcDNA3.1-GPX4 was determined using western blot. (B) GPX4 overexpression restored cell proliferation which was inhibited by rhamnazin in SMMC-7721 and Huh-7 cells. (C) GPX4 overexpression restored colony formation which was inhibited by rhamnazin in SMMC-7721 and Huh-7 cells. (D-F) Reversal of ferroptosis by GPX4 overexpression in rhamnazin-induced HCC cells. **p < 0.01 compared with rhamnazin (10 μM) treatment group.
The purpose of traditional anti-carcinoma therapies is to promote carcinoma cell apoptosis, but a wide variety of cancer cells are frequently defective in apoptosis inducers or chemo-resistant. Hence, exploring new agents that induce non-apoptotic forms of cell death will offer potential therapeutic candidates for malignancies. Ferroptosis is a newly discovered type of regulated cell death (Su et al. 2020). Characterized by Fe2+ overload and lipid peroxidation, ferroptosis has been broadly reported to be involved in HCC (Zhang et al. 2019). Recent studies have demonstrated that suppressing ferroptosis might be a pivotal signal for liver cancer initiation, thus providing a new way to combat liver cancer (Zhang et al. 2018; Chen et al. 2019). Small pharmacological inhibitors, including the GPX4 inhibitor (1S, 3R)-RSL3 (RSL3) and erastin as direct inhibitors of xCT‐mediated import function, are widely used for the induction of ferroptosis (Dixon et al. 2012; Gao et al. 2021). Accumulating evidence indicates that ferroptosis inhibitors have antioncogenic potentials in multiple tumor types, including liver cancer (Louandre et al. 2013; Chen et al. 2020a; Yang et al. 2021).
As an inhibitor of multiple oncogenic kinases, sorafenib is an approved systemic therapy for advanced HCC (Lachaier et al. 2014). Growing evidence has confirmed that sorafenib inhibits HCC cells through the induction of ferroptosis rather than apoptosis (Dixon et al. 2014; Sun et al. 2021a). Recently, rhamnazin increases the chemotherapeutic effect of sorafenib in HCC cells has been documented (Habiba et al. 2022). Analogous to small molecule kinase inhibitors, rhamnazin inhibits VEGF-induced phosphorylation of VEGFR2 and tumor angiogenesis (Yu et al. 2015). However, much is still not known about the roles of rhamnazin in HCC cells ferroptosis. In this work, we disclosed for the first time that rhamnazin exerted its anti-cancer action via causing ferroptosis and preventing cell growth and invasion in HCC cells.
In cell proliferation assay, rhamnazin-induced HCC cell growth inhibition was not reversed by Z-VAD-FMK, CQ, or necrostatin-1, suggesting that another type of cell death might have occurred. Accordingly, we sought to explore whether treatment with rhamnazin-induced the ferroptosis in HCC cells. The ferroptosis-related events, including ROS accumulation, iron content, and lipid peroxidation, were reinforced by rhamnazin. To determine how rhamnazin induces ferroptosis, the intrinsic or enzyme-regulated pathway and the extrinsic or transporter-dependent pathway, which initiates ferroptosis, were evaluated. Substantial evidence manifested that rhamnazin interfered with the intrinsic pathway rather than the extrinsic pathway. The GPX4 protein expression was sharply declined by rhamnazin in a dose-dependent manner. Due to the limitation of technical tools, we could not investigate the potential binding modes of rhamnazin and GPX4 using AutoDock software. In the follow-up study, we will evaluate the binding sites of rhamnazin within GPX4. Moreover, in rescue assay, pretreatment with the GPX4 overexpression plasmid reduced rhamnazin-induced HCC cell ferroptosis.
Ferroptosis event also contributes to the anti-cancer influence of multiple tumor suppressor genes, including BRCA1-associated protein 1 (BAP1) and p53 (Lin et al. 2020). The p53-mediated transcriptional inhibition of SLC7A11 contributed to ROS-induced ferroptosis (Xie et al. 2017). Hence, whether rhamnazin regulates the ferroptosis of HCC cells by affecting p53 deserves further exploration. The combination treatment of sorafenib and compound exhibits a notable synergistic anticancer effect (Li et al. 2019). Whether the synergy between rhamnazin and sorafenib in inducing ferroptosis warrants further investigation. Taking into consideration that the precise mechanism underlying rhamnazin causes ferroptosis require further investigation, the follow-up studies about combination effect of rhamnazin and sorafenib will be conducted in the future. We are currently searching for suitable parameters to effectively evaluate the synergistic or antagonistic effect of the combination. Another deficiency of this study is the absence of evaluation the in vivo antitumor effect of rhamnazin, which would hinder its translation to clinical applications. In the future, we will adopt HCC patient-derived xenograft (PDX) model and verified the tumor-inhibiting role of rhamnazin in vivo.
Genetic or pharmacologic ferroptosis induction reverses sorafenib-resistance in HCC (Sun et al. 2016; Gao et al. 2021). Therefore, we speculated that rhamnazin was also able to efficiently reverse the sorafenib resistance effect of HCC via inducing ferroptosis. More mechanisms of HCC sorafenib resistance reversed by rhamnazin require further investigation. Rhamnazin is expected to have substantial utility in providing a concomitant effect with existing drugs (sorafenib) or providing a greater variety of pharmacological treatment options for HCC. Inhibition of VEGFR signaling pathway has already become one of the most promising approaches for the treatment of cancer. VEGFRs contain multiple phosphorylation sites and active pockets. Rhamnazin inhibits VEGFR2 phosphorylation at Tyr951, while sorafenib inhibits VEGFR2 phosphorylation at Tyr1054 (Wilhelm et al. 2004; Yu et al. 2015). In anti-angiogenesis treatment, the antagonistic or synergistic effects of the combination sorafenib and rhamnazin will be investigated in the following work.
Immunotherapy-activated CD8+ T cells enhance ferroptosis-specific lipid peroxidation in tumor cells, and in turn, increases ferroptosis contributes to the anti-tumor efficacy of immunotherapy (Wang et al. 2019; Sun et al. 2021b). The key question for follow-up studies to identify the exacerbated immune response driven by rhamnazin in HCC has important clinical implications. Several studies have established a correlation between the VEGF-VEGFR2 axis and an immunosuppressive microenvironment; this immunosuppression can be overcome by anti-angiogenic reagents (Yasuda et al. 2013; Tada et al. 2018). Being an inhibitor of VEGFR2, whether or not, tumor immune microenvironment is regulated by rhamnazin is also well worth to further study (Yu et al. 2015).
Collectively, this study provided novel insights that rhamnazin might be a ferroptosis-inducing agent and an effective anti-cancerous compound in HCC cells (Fig. 6).

A proposed model showing that rhamnazin induces ferroptosis in HCC cells.
The authors declare no conflict of interest.