Abstract
Cancer is a clonal disease that develops as a result of the changes on the genetic material by various factors in micro/macro environment. It has a multi-step development process. In some cancer types, genetic factors allow this multi-step process to proceed easily. These cancer types are also called hereditary cancer syndromes. Targeted gene panels are important diagnostic methods in hereditary cancer syndromes to detect the causative variants associated with these hereditary cancer syndromes. We reviewed the data of 94 patients who applied to Ankara City Hospital Genetic Diseases Evaluation Center from March 2019 to July 2021. Qiagen familial cancer susceptibility gene panel kit was used for next generation sequencing to detect the single nucleotide variants for the targeted genes. Sixty-one genes which are associated with increased cancer risk or well characterized hereditary cancer syndromes were included to this panel. Twenty five patients (27%), including 8 males and 17 females, had pathogenic/likely pathogenic variants in 13 of the 61 genes analyzed. Forty patients (43%) had variants which were assessed as variant of unknown significant. In our study, targeted multi-gene panel was diagnostic in nearly one third of the patients with personal/familial cancer syndromes. Molecular diagnosis in familial cancer syndromes is important in terms of predictive diagnosis and family screening, as well as patient follow-up and early prophylactic surgery. The predisposition for hereditary cancer syndromes can be determined according to pre-test evaluation, figuring out the inheritance type with pedigree analysis, cancer type and the genetic analysis for appropriate susceptibility genes.
Introduction
Cancer is a clonal disease that develops as a result of the changes on the genetic material by various factors in micro/macro environment with a multi-step development process. In some cancer types, genetic factors allow this multi-step process to begin easily. In this case, it is noted that there is a “genetic predisposition” for cancer. These cancer types are also referred to as hereditary cancer syndromes. Hereditary cancers make up 5-10% of all cancer types. Most of them are inherited as autosomal dominant (American College of Obstetricians and Gynecologists 2019). Up to now, more than 200 hereditary cancer syndromes have been reported (Machačkova et al. 2016; Tsaousis et al. 2019).
Targeted gene panels are important diagnostic methods in hereditary cancer syndromes (LaDuca et al. 2014). There are many advantages of targeted next generation sequencing (NGS) gene panels utilization in hereditary cancer syndromes, including cost-effectiveness or time saving (Pritchard et al. 2012; LaDuca et al. 2014). Morbidity and mortality can be reduced by the correct genetic diagnosis in patients with hereditary cancer syndromes and the preventive procedures can be performed to the patients. The family members that are at risk can also be tested, and genetic counseling can be given to them (according to the test results) (de Oliveira et al. 2022).
This study aims to detect the frequency of germline pathogenic/likely pathogenic variants in cancer susceptibility genes by the utilization of targeted NGS panels for the patients or family members referred with hereditary cancer syndromes.
Materials and Methods
Patients
We reviewed the data of 94 patients as two groups who applied to Ankara City Hospital Genetic Diseases Evaluation Center from March 2019 to July 2021. One group of patients had a history of different solid tumors affecting more than one system in themselves and/or their families, while the other group underwent BRCA1/2 deletion/duplication analysis for breast-ovarian cancer and had no pathogenicity. The patients were referred to our genetic laboratory for diagnostic genetic test.
Permission for the study was obtained from Ankara City Hospital Ethics Committee (E1-22-2488). The study followed the guidelines and principles of the Declaration of Helsinki. All patients and formal guardians of the patients under 18 signed the written informed consent for the usage of their clinical data and genetic analysis.
Genomic DNA was extracted from peripheral blood using QIAcube® automatic DNA isolation system (QIAGEN Inc., Mississauga, ON, Canada) according to the manufacturer’s instructions. Qiagen familial cancer susceptibility gene panel CDHS-13974Z-2393 kit (QIAGEN, Hilden, Germany) was used for NGS to detect the single nucleotide variants for the targeted genes. The target enrichment process was followed by sequencing of the libraries on Illumina MiSeq sysytem (Illumina Inc., San Diego, CA, USA).
Data analysis and variant interpretation
Data analysis was carried out by QIAGEN Clinical Insight (QCITM) software (QIAGEN). Pathogenic, likely pathogenic, and uncertain significant variants were confirmed by Sanger sequencing. The exons of all targeted genes were sequenced at a read depth of 30 × or greater. 2015 American College of Medical Genetics Standards and Guidelines (ACMG) were used for the interpretation of sequence variants 10 (Richards et al. 2015).
Results
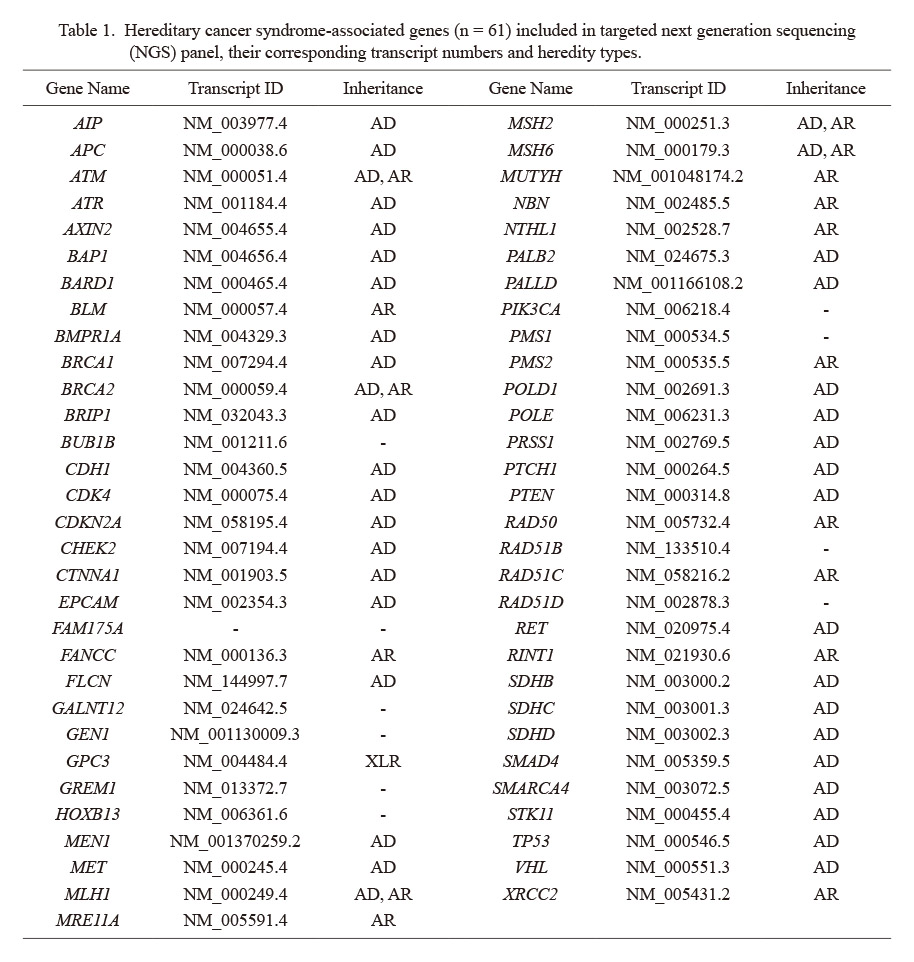
A total of 94 patients (33 males and 61 females) with suspected hereditary cancer syndromes were analysed for molecular etiology. The range of ages of the patients was between 2-71. Thirty three male cases had a mean age of 45.7; and 61 female cases had a mean age of 40.2. Sixteen patients were in the pediatric age group. Sixty-one genes which are associated with increased cancer risk or well characterized hereditary cancer syndromes were included to this panel (Table 1).
Out of 94 cases referred to us for genetic testing, 90 patients (96%) had a personal cancer history. The majority of the patients had a personal history of colon adenocarcinoma (14%) and breast cancer (14%). A family history for cancer was positive for 49% (46/94) of the whole patients. Pathogenic variants were detected in 21 of these 46 patients with a positive family history, and variant of unknown significant (VUS) were detected in 19 of them, six patients had no pathogenic variant/VUS. Missense variants were the most prevalent variation type, accounting for the 40% (10/25) of the variants identified, followed by nonsense, frameshift, insertion/deletion variations and splicing variations.
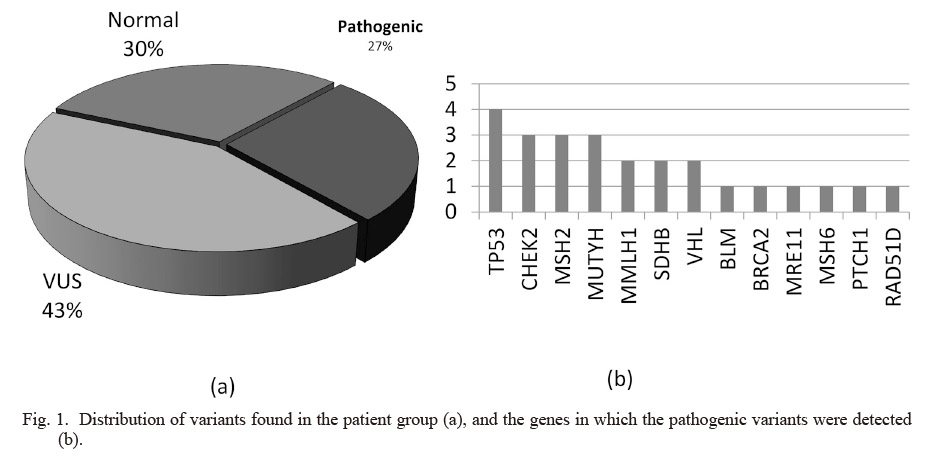
Twenty five patients (27%), including 8 males and 17 females, had pathogenic/likely pathogenic variants in 13 of the 61 genes analyzed (TP53, CHEK2, MSH2, MUTYH, MMLH1, SDHB, VHL, BLM, BRCA2, MRE11, MSH6, PTCH1 and RAD51D genes) (Fig. 1a). Five patients (19%) were under 18 years old. Family history was positive at twenty two patients out of twenty five patients who had pathogenic/likely pathogenic variants. Only two patients had no positive family history, and both of them had pathogenic variant at SDHB gene that has autosomal dominant inheritance. While one of these patients had paraganglioma, the other had pheochromocytoma. The other pathogenic variants were at TP53 (4), CHEK2 (3), MSH2 (3), MUTYH (3), MMLH1 (2), VHL (2), BLM (1), BRCA2 (1), MRE11 (1), MSH6 (1), PTCH1 (1) and RAD51D (1) genes (Fig. 1b). One patient diagnosed with colon cancer had compound heterozygosity for MUTYH gene. Another patient diagnosed with lymphoblastic lymphoma had homozygote pathogenic variant at MSH2 gene. The family histories, ages, and sex of the patients, genes and pathogenic/likely pathogenic variants are shown in Table 2.
BRCA1/2 MLPA (Multiplex Ligation-dependent Probe Amplification) were only performed on seven patients with breast cancer, three patients with ovarian cancer and one patient with pancreas cancer because of the high frequency of BRCA deletion/duplications in these cancer types. No variants were found. With targeted gene NGS, two patients had pathogenic variants at TP53 and RAD51D genes, four patients had VUS at APC, PALB2, RAD50 and PTCH1 genes. Five patients had no variations.
Forty patients (43%) had variants which were assessed as variant of unknown significant in our study (Fig. 1a). VUS was detected at NBN (1), PMS1 (2), BUB1B (2), CTNNA1 (1), SDHD (2), ATR (4), STK11 (1), RAD50 (3), POLE (3), RET (1), GEN1 (1), PTCH1 (2), FLCN (1), ATM (2), RAD51D (1), MSH6 (1), POLD1 (2), NTHL1 (1), PALB2 (2), SMARCA4 (2), CDH1 (2), MRE11 (2), BRIP1 (1), PMS2 (1), BARD1 (1), RAD51C (1) and APC (2) genes. Two patients had two and three VUS in different genes. These patients were followed up and called for annual controls. Pathogenic, likely pathogenic and VUS were confirmed by bidirectional Sanger sequencing. Over 99% of the coding exons of all genes in the panel were sequenced to a read depth of 30 × or greater in almost all cases. According to these results, molecular diagnosis rate was 27%.
Discussion
Multi-gene panels using NGS technology make it possible to identify pathogenic variants at hereditary cancer susceptibility genes. Each mutated gene included in the panel can increase the risk for hereditary cancer syndomes (de Oliveira et al. 2022). Thus, these multi-gene panels ensure the risk estimation in predictive medicine (Colas et al. 2019). In this study, hereditary cancer related pathogenic variants were detected by using NGS technology at the susceptibility genes for hereditary cancers. A total of 94 individuals who were referred to our clinics for hereditary cancer syndromes were included in our study. All of the individuals had personal/family history. The genetic analysis disclosed the presence of at least one pathogenic/likely pathogenic variant in 27% of the individuals, and VUS were detected in 43% of the cases. Thus, the contribution of all genes in our panel was important for the hereditary cancer predisposition. The diagnosis rate was similar to previous studies (Tsaousis et al. 2019; Ercoskun et al. 2022).
Pathogenic variants were detected mostly at patients with colon adenocarcinoma (24%), following ovarian cancer (12%), brain tumors (12%), breast cancer (8%), paraganglioma (8%) and less at patients with other types of cancer. Among the patients with a pathogenic variant, 16% (4/25) of the cases had variants in the TP53 gene, indicating a significant contribution of this gene in cancer predisposition. Apart from the TP53 gene, the other highly mutated genes were CHEK2 (12%), MUTYH (8% monoallelic variants, 4% biallelic variants), and MSH2 (12%). At least one pathogenic/likely pathogenic variant was identified in 25 of samples (27%) including all of the individuals with no information about their personal/family cancer history.
The rate of hereditary cancer syndromes in adults is between 5-10%, but in children it has not been fully investigated. In a study by Knapke et al. (2012) at a pediatric cancer clinic, the patients were followed for over 2-years. They were screened for family history, age, ethnicity and tumor characteristics underlying cancer predisposition syndrome. Pathogenic variants were detected at 29% of the children due to the family history with cancer (Knapke et al. 2012; Schiffman et al. 2013). In our study, the diagnosis rate in pediatric age group was 37.5%, while it was 24% in adulthood period, compatible with the literature.
Biallelic pathogenic variants of MUTYH gene are associated with a syndrome known as MUTYH associated polyposis (MAP) syndrome (Tsaousis et al. 2019). In only one case (a 66 years old female patient with colon adenocarcinoma), MUTYH variations were detected as compound heterozygote in our study. Monoallelic MUTYH pathogenic variant is also significant as a risk factor for colorectal cancer. The risk is 2.5 times more according to general population (Win et al. 2014). Two cases in our study were heterozygote for different monoallelic variations of MUTYH gene. One of the patients was diagnosed with breast cancer and the other had recurrent endometrial polyp with positive family histories, respectively. MUTYH heterozygotes may have a mildly elevated risk for colorectal carcinoma according to NCCN guidelines (NCCN, The National Comprehensive Cancer Network 2018). There was no other multiple pathogenic variant found in any of our patients except from MUTYH.
Pathogenic variants of TP53 are associated with a wide range of cancers, collectively known as Li Fraumeni Syndrome, which is characterised by a predisposition to some malignancies like adrenocortical carcinomas, soft-tissue sarcomas, early-onset breast cancer, brain tumors and leukaemias (Paduano et al. 2021, 2022). TP53 had the highest rate (16%) for pathogenic variant in our study. The patients had different cancer types (adrenocortical carcinoma, brain tumor, breast and colon cancer) with positive family history, in line with the literature.
SDHB pathogenic variants were detected in two patients with paraganglioma in our study. Succinate dehydrogenase genes (SDHA, SDHB, SDHC, SDHD, and SDHAF2) are associated with hereditary paraganglioma syndromes in literature. For paraganglioma syndromes, to identify a pathogenic variant is important for the further surveillance protocol recommendations to the patients and their families. This actually illustrates the importance of analyzing susceptibility gene variants in familial cancer syndromes and the importance of the diversity of the genes included in the panel for different familial cancer types (Horton et al. 2022).
A pathogenic missense variant in BLM gene was detected at 44 years old female patient who was diagnosed with colon adenocarcinoma. Her sister had renal cell carcinoma, her cousin had lung adenocarcinoma and her grandmother had pancreas cancer. de Voer et al. (2015) reported two patients carried a pathogenic BLM variant in a cohort of 55 early-onset colorectal carcimoma cases (≤ 45 years old). Pathogenic BLM variations may cause an increased risk for developing colorectal carcinoma at an early age, but some other risk factors are also important for the carcinogenesis period (de Voer et al. 2015).
The overall rate of VUS in our study was around 43-45% similar to other studies with a maximum of two VUS per individual (Tsaousis et al. 2019; Ercoskun et al. 2022). The highest VUS rate among individuals with personal history of cancer was detected with breast cancer (12.5%), colon adenocarcinoma (10.0%) and pancreas cancer (10.0%). Only one unaffected individual with family history of ovarian cancer had VUS at CDH1 gene. Family study may also be recommended at VUS variants, but it may not provide complete information because of the incomplete penetrance and variable expression of cancer susceptibility (Garrett et al. 2016; Anderson et al. 2022). Patients with VUS were called for annual controls. Since the age of onset is different in patients, it is appropriate to evaluate the patients with VUS once a year (Colas et al. 2019). This is because accurate VUS assessment needs a professional review of literature using ACMG guidelines (Anderson et al. 2022). As the data gets bigger, specific revisions for disease or the variants are required (Nykamp et al. 2017; Waddell-Smith et al. 2020) In addition to this, as the number of genes included in the panel increases, there will be a higher VUS risk (de Oliveira et al. 2022).
Molecular diagnosis in familial cancer syndromes is important in terms of predictive diagnosis and family screening, as well as patient follow-up and early prophylactic surgery. Selecting panels containing target genes will enable patients to receive molecular diagnosis in a shorter time. Studying large multigene panels will contribute to the diagnosis in cases with atypical phenotypic features or when the etiology cannot be clarified with single gene-targeted studies. It is also recommended that laboratories performing multiple gene testing share their datas, thereby contributing to the increase of this useful information.
In our study, targeted multi-gene panel was diagnostic in nearly one third of the patients with personal/familial cancer syndromes. The predisposition for hereditary cancer syndromes can be determined according to pre-test evaluation, deciding the inheritance type with pedigree analysis, cancer type and the genetic analysis for appropriate susceptibility genes. Variable onset of the disease, personal cancer or positive family history are important for a correct genetic diagnosis. An accurate diagnosis is also important for an appropriate genetic counseling for the patients to understand the significance of genetic testing.
Conflict of Interest
The authors declare no conflict of interest.
References
-
American College of Obstetricians and Gynecologists (ACOG)
(2019) Hereditary cancer syndromes and risk assessment: ACOG COMMITTEE OPINION, number 793. Obstet. Gynecol., 134, e143-e149.
-
Anderson,
C.L.,
Munawar,
S.,
Reilly,
L.,
Kamp,
T.J.,
January,
C.T.,
Delisle,
B.P. &
Eckhardt,
L.L.
(2022) How functional genomics can keep pace with VUS identification. Front. Cardiovasc. Med., 9, 900431.
-
Colas,
C.,
Golmard,
L.,
de Pauw,
A.,
Caputo,
S.M. &
Stoppa-Lyonnet,
D.
(2019) “Decoding hereditary breast cancer” benefits and questions from multigene panel testing. Breast, 45, 29-35.
-
de Oliveira,
J.M.,
Zurro,
N.B.,
Coelho,
A.V.C.,
Caraciolo,
M.P.,
de Alexandre,
R.B.,
Cervato,
M.C.,
Minillo,
R.M.,
de Vasconcelos Carvalho Neto,
G.,
Grivicich,
I. &
Oliveira,
J.B.
(2022) The genetics of hereditary cancer risk syndromes in Brazil: a comprehensive analysis of 1682 patients. Eur. J. Hum. Genet., 30, 818-823.
-
de Voer,
R.M.,
Hahn,
M.M.,
Mensenkamp,
A.R.,
Hoischen,
A.,
Gilissen,
C.,
Henkes,
A.,
Spruijt,
L.,
van Zelst-Stams,
W.A.,
Kets,
C.M.,
Verwiel,
E.T.,
Nagtegaal,
I.D.,
Schackert,
H.K.,
van Kessel,
A.G.,
Hoogerbrugge,
N.,
Ligtenberg,
M.J.,
et al.
(2015) Deleterious germline BLM mutations and the risk for early-onset colorectal cancer. Sci. Rep., 5, 14060.
-
Ercoskun,
P.,
Yuce Kahraman,
C.,
Ozkan,
G. &
Tatar,
A.
(2022) Genetic characterization of hereditary cancer syndromes based on targeted next-generation sequencing. Mol. Syndromol., 13, 123-131.
-
Garrett,
L.T.,
Hickman,
N.,
Jacobson,
A.,
Bennett,
R.L.,
Amendola,
L.M.,
Rosenthal,
E.A. &
Shirts,
B.H.
(2016) Family studies for classification of variants of uncertain classification: current laboratory clinical practice and a new web-based educational tool. J. Genet. Couns., 25, 1146-1156.
-
Horton,
C.,
LaDuca,
H.,
Deckman,
A.,
Durda,
K.,
Jackson,
M.,
Richardson,
M.E.,
Tian,
Y.,
Yussuf,
A.,
Jasperson,
K. &
Else,
T.
(2022) Universal germline panel testing for individuals with pheochromocytoma and paraganglioma produces high diagnostic yield. J. Clin. Endocrinol. Metab., 107, e1917-e1923.
-
Knapke,
S.,
Nagarajan,
R.,
Correll,
J.,
Kent,
D. &
Burns,
K.
(2012) Hereditary cancer risk assessment in a pediatric oncology follow-up clinic. Pediatr. Blood Cancer, 58, 85-89.
-
LaDuca,
H.,
Stuenkel,
A.J.,
Dolinsky,
J.S.,
Keiles,
S.,
Tandy,
S.,
Pesaran,
T.,
Chen,
E.,
Gau,
C.L.,
Palmaer,
E.,
Shoaepour,
K.,
Shah,
D.,
Speare,
V.,
Gandomi,
S. &
Chao,
E.
(2014) Utilization of multigene panels in hereditary cancer predisposition testing: analysis of more than 2,000 patients. Genet. Med., 16, 830-837.
-
Machačkova,
E.,
Hazova,
J.,
Stahlova Hrabincova,
E.,
Vasickova,
P.,
Navratilova,
M.,
Svoboda,
M. &
Foretova,
L.
(2016) Retrospective NGS study in high-risk hereditary cancer patients at Masaryk Memorial Cancer Institute. Klin. Onkol., 29 Suppl 1, S35-45.
-
Nykamp,
K.,
Anderson,
M.,
Powers,
M.,
Garcia,
J.,
Herrera,
B.,
Ho,
Y.Y.,
Kobayashi,
Y.,
Patil,
N.,
Thusberg,
J.,
Westbrook, M. & Topper, S; Invitae Clinical Genomics Group
(2017) Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria. Genet. Med., 19, 1105-1117.
-
Paduano,
F.,
Colao,
E.,
Fabiani,
F.,
Rocca,
V.,
Dinatolo,
F.,
Dattola,
A.,
D’Antona,
L.,
Amato,
R.,
Trapasso,
F.,
Baudi,
F.,
Perrotti,
N. &
Iuliano,
R.
(2022) Germline testing in a cohort of patients at high risk of hereditary cancer predisposition syndromes: first two-year results from South Italy. Genes (Basel), 13, 1286.
-
Paduano,
F.,
Fabiani,
F.,
Colao,
E.,
Trapasso,
F.,
Perrotti,
N.,
Barbieri,
V.,
Baudi,
F. &
Iuliano,
R.
(2021) Case report: identification of a novel pathogenic germline TP53 variant in a family with Li-Fraumeni syndrome. Front. Genet., 12, 734809.
-
Pritchard,
C.C.,
Smith,
C.,
Salipante,
S.J.,
Lee,
M.K.,
Thornton,
A.M.,
Nord,
A.S.,
Gulden,
C.,
Kupfer,
S.S.,
Swisher,
E.M.,
Bennett,
R.L.,
Novetsky,
A.P.,
Jarvik,
G.P.,
Olopade,
O.I.,
Goodfellow,
P.J.,
King,
M.C.,
et al. (2012) ColoSeq provides comprehensive lynch and polyposis syndrome mutational analysis using massively parallel sequencing. J. Mol. Diagn., 14, 357-366.
-
Richards,
S.,
Aziz,
N.,
Bale,
S.,
Bick,
D.,
Das,
S.,
Gastier-Foster,
J.,
Grody,
W.W.,
Hegde,
M.,
Lyon,
E.,
Spector,
E.,
Voelkerding, K. & Rehm, H.L.; ACMG Labolatory Quality Assurance Committee
(2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med., 17, 405-424.
-
Schiffman,
J.D.,
Geller,
J.I.,
Mundt,
E.,
Means,
A.,
Means,
L. &
Means,
V.
(2013) Update on pediatric cancer predisposition syndromes. Pediatr. Blood Cancer, 60, 1247-1252.
-
The National Comprehensive Cancer Network (NCCN)
(2018) Genetic/Familiar High-Risk Assessment: Colorectal, Version 1.2018. Inherited Variants at Hereditary Cancer Syndromes 7 UNCORRECTED PROOF. https://www.nccn.org/professionals/physician_gls/pdf/genetics_colon.pdf [Accessed: August 27, 2018].
-
Tsaousis,
G.N.,
Papadopoulou,
E.,
Apessos,
A.,
Agiannitopoulos,
K.,
Pepe,
G.,
Kampouri,
S.,
Diamantopoulos,
N.,
Floros,
T.,
Iosifidou,
R.,
Katopodi,
O.,
Koumarianou,
A.,
Markopoulos,
C.,
Papazisis,
K.,
Venizelos,
V.,
Xanthakis,
I.,
et al. (2019) Analysis of hereditary cancer syndromes by using a panel of genes: novel and multiple pathogenic mutations. BMC Cancer, 19, 535.
-
Waddell-Smith,
K.E.,
Skinner,
J.R. &
Bos,
J.M.
(2020) Pre-test probability and genes and variants of uncertain significance in familial long QT syndrome. Heart Lung Circ., 29, 512-519.
-
Win,
A.K.,
Dowty,
J.G.,
Cleary,
S.P.,
Kim,
H.,
Buchanan,
D.D.,
Young,
J.P.,
Clendenning,
M.,
Rosty,
C.,
MacInnis,
R.J.,
Giles,
G.G.,
Boussioutas,
A.,
Macrae,
F.A.,
Parry,
S.,
Goldblatt,
J.,
Baron,
J.A.,
et al. (2014) Risk of colorectal cancer for carriers of mutations in MUTYH, with and without a family history of cancer. Gastroenterology, 146, 1208-1211. e1201-1205.