Regular Contribution
Clinical Evaluation of Pediatric Patients with Hereditary Angioedema
2024 Volume 262 Issue 1 Pages 23-27

Details
2024 Volume 262 Issue 1 Pages 23-27
Hereditary angioedema is a rare, potentially life-threatening disease. There is a lack of data describing the clinical course of hereditary angioedema (HAE) in children. We aimed to evaluate the clinical characteristics of pediatric patients with hereditary angioedema: The age of disease onset, age at diagnosis, the frequency of angioedema attacks, the total number of attacks before diagnosis, the regions where angioedema attacks were observed, accompanying abdominal pain, and serum levels of C4 and C1 esterase inhibitor were obtained and recorded. In addition, the results of SERPING1 (C1INH) gene sequence analysis of the patients in this group were also collected from medical records and recorded. While none of the patients reported a skin rash as a symptom of attack, there was formication observed in the region of angioedema in 46.9% (n = 15) of the patients and pruritus in 6.2% (n = 2) of the patients. At disease onset, the complaints of the patients regarding location of edema were on the hands of 32.3% (n = 10), on the feet of 9.7% (n = 3), on the faces of 25.7% (n = 8), and abdominal attacks in 32.3% of the patients (n = 10). Four different variants, one of which was novel, were detected in the SERPING1 gene in eight different families. The results of this study suggest that hereditary angioedema is diagnosed only when the patient requests examination following recurrent angioedema. Severe laryngeal edema attacks in patients without a diagnosis of HAE are fatal at a higher rate than attacks in patients with a diagnosis. Thus, awareness of the symptoms of HAE is necessary, and correct diagnosis is essential to proper treatment.
Hereditary angioedema (HAE) is a rare autosomal dominant disease that results from the deficiency or dysfunction of C1 esterase inhibitor (C1-INH) (Nzeako et al. 2001). Heterozygous variants in the C1 inhibitor (C1NH; SERPING1) gene were identified as causative for HAE1 and HAE2 (Santacroce et al. 2021). Autosomal recessive inheritance has also been reported (Blanch et al. 2006). Moreover, the genetic etiology of HAE is heterogeneous, and HAE with normal C1-INH has been associated with various genes including F12, PLG, ANGPT1, KNG1, MYOF, and HS3ST6 (Santacroce et al. 2021). The exact prevalence of the disease, which has been observed equally in both sexes, is not known. However, it has been accepted to be in the proportion of 1/50,000-100,000 (Agostoni et al. 2004; Bowen et al. 2008). Three phenotypes of HAE have been determined. While the low serum levels of C1-INH are the cause of angioedema attacks in Type 1 HAE, dysfunction of C1-INH causes angioedema attacks in type 2 HAE despite normal-to-high serum levels. In type 3 HAE, both levels and function of C1-INH are normal, yet HAE symptoms are often observed. Angioedema attacks might affect any region of the body but they occur most frequently on faces, tongue, visceral organs, and extremities. Angioedema attacks begin before the age of 10 in half of patients with HAE, whose onset is very rare in the infantile period. The clinical course of the disease worsens during adolescence in many patients. In pediatric patients, it has been observed most commonly on the face, body, genital regions, and in extremities of pediatric patients.
Subcutaneous edema develops without accompanying erythema and pruritus (Frank et al. 1976; Burks et al. 2020). Mechanical trauma is the most important factor causing the onset of attacks in children (Farkas et al. 2017). Swelling that develops in HAE attack reaches its largest size in 24 hours. HAE displays a slow course of recovery within the following 48-72 hours, and it ends within approximately 72-96 hours. The abdominal complaints related with HAE depend on the edema in the gastrointestinal system, and the developed abdominal pain is mostly incorrectly diagnosed as acute abdominal pain or familial Mediterranean fever. Checking serum C1 esterase inhibitor levels and function not only provides the diagnosis but also enables the physician to make the distinction between the types of HAE. Edemas with mild attacks generally regress spontaneously but fresh frozen plasma and the recombinant C1 esterase inhibitor, icatibant treatment, have been used to replace degraded C1 esterase inhibitor if severe edema exist in the patient. The plasma kallikrein inhibitor ecallantide is recommended for use in adolescents but not approved for use under 12 years of age (Maurer et al. 2022). Training the patient and their family is very important in the treatment of HAE (Ozkars et al. 2019; Maurer et al. 2022). There are a limited number of studies about the clinical course of HAE in pediatric patients. In this study, we aimed to evaluate the clinical characteristics, particularly the age at disease onset, the age at diagnosis, and the number of attacks until the time of diagnosis of pediatric patients with HAE.
The patients who were diagnosed with hereditary angioedema in the Pediatric Allergy Polyclinic of XXXX XXX Hospital between July 1, 2019 and April 1, 2021 were included in this study. Permission was obtained from the patient to use his/her photograph for the graphical abstract. The age of disease onset, age at diagnosis, the frequency of angioedema attacks, the total number of attacks before diagnosis, the regions where angioedema attacks were observed, accompanying abdominal pain, and serum levels of C4 and C1 esterase inhibitor were obtained and recorded. In addition, the results of SERPING1 (C1INH) gene sequence analysis in this group were also collected from medical records.
The diagnosis of hereditary angioedema was made if the protein levels of C1 esterase inhibitor were low or if activity levels were low (Maurer et al. 2022). Levels of C1 esterase inhibitor less than 0.21 g/L and levels of C4 less than 10 mg/dL were accepted as low.
Molecular studies were performed on 22 of 32 patients diagnosed with HAE based on clinical findings. Genomic DNA was extracted from the peripheral blood leukocytes of the patients using an PureLink Genomic DNA Mini Kit (Invitrogen, Carlsbad, CA, USA), in accordance with the manufacturer’s instructions. PCR reactions were performed with in-house primers designed specifically for the exonic regions of the SERPING1 gene. Next generation sequencing analysis for all coding exons and exon-intron boundaries (+/–10) of SERPING1 was performed by using MGI FS DNA Library kit and MGI DNBSEQ-G400. The DNA sequences were aligned to the GRCh37 (hg19) human reference genome and evaluated with Integrated Genomic Viewer software. Genome Aggregation Database (gnomAD), 1000 genome project, and dbSNP data were used as the control population. The detected variants were analyzed using in silico pathogenicity prediction tools such as SIFT, Polyphen, and MutationTaster. The identified variants were classified according to the American College of Medical Genetics and Genomics (ACMG) guidelines (Richards et al. 2015). The approval of XXXX University Clinical Researches Ethics Committee was obtained.
There was a homogenous distribution in the study in terms of female and male patients. The presence of other patients with angioedema in the family was found in 29 (90.6%) patients. The mean age of the patients was 9.9 ± 5.2 years [mean ± standard deviation (SD)]. The age of onset of angioedema was 6.6 ± 4.0 years, while the age at diagnosis with angioedema was 9.7 ± 4.9 years. The average number of attacks per patient per year was determined as 38.2 ± 31.2. It was also found that the total number of attacks until the time of diagnosis was 88.3 ± 80.1. The C4 level was above 10 mg/dL in 2 of patients (6.2%) and C1-INH activity level was normal in 5 patients (15.6%). C1-INH activity level was found low in these patients.
When the regions where angioedema attacks occurred were considered, it was observed that they occurred mostly on the hands in 29 patients (90.6%), while abdominal attacks were observed in 23 patients (71.8%).
While no patients reported skin rash as messenger attacks, 15 (46.9%) reported formication in the angioedema region, and 2 patients (6.2%) reported pruritus in the region. As for the regions where there were complaints at the onset of the disease, 10 patients (32.3%) had problem on their hands, 3 (9.7%) on their feet, 8 (25.7%) on their face, and 10 (32.3%) on their abdomen (Table 1).
Four different variants, one of which was novel, were detected in eight different families in the SERPING1 (NM_000062.3) gene. The variant c.1033G>T (p.Gly345Trp) was detected in sixteen individuals from five families. This variant was classified as likely pathogenic according to the ACMG criteria (PM1, PM5, PP3, PM2) and has been previously associated with the disease in the literature (Ozkars et al. 2019). The variant c.1438_1442delGTGCTinsATGCC (p.Val480_Leu481delinsMetPro) was identified in three members of a family. This variant was classified as uncertain according to the ACMG criteria (PM1, PM2). It was not found in gnomAD genomes. The variant c.449C>T (p.Ser150Phe) was detected in two members of a family, which was previously reported in the literature (Yasuno et al. 2021). This variant has been interpreted as likely pathogenic according to the ACMG criteria (PP3, PM1, PM5, PM2) and predicted as deleterious by the PolyPhen-2 (with a score of 0.968) and SIFT programs. The variant c.1397G>A (p.Arg466His), which has been previously associated with the disease, was detected in one patient (Rijavec et al. 2013) (Table 2). The identified variant was classified as pathogenic based on ACMG variant classification guidelines (PP5, PM1, PM5, PP3, PM2) and predicted to be disease causing by in silico pathogenicity prediction tools such as MutationTaster and SIFT.

Statistical results of patient data.
C1-INH, C1 esterase inhibitor.
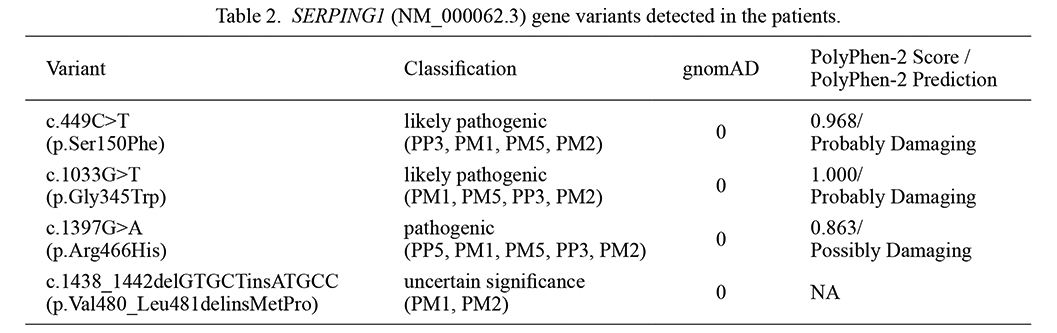
SERPING1 (NM_000062.3) gene variants detected in the patients.
gnomAD, The Genome Aggregation Database; NA, not available.
This study was carried out to evaluate pediatric patients with hereditary angioedema clinically since it is a disease that is rarely seen in children. The age of disease onset, the age at diagnosis, the frequency of angioedema attacks, the total number of attacks before diagnosis, the regions where angioedema attacks are observed, accompanying abdominal pain, the C4 and C1 esterase inhibitor levels, and the results of SERPING1 gene sequence analysis of the patients were all evaluated.
There are significant differences in prodromal symptoms that can occur before or during the attacks of HAE. Erythema marginatum can be observed in some patients with hereditary angioedema before the attacks. In the related literature, there are some publications that claim it can be seen in 26% to 56% of patients. Conversely, other publications expressed that it was never observed (Agostoni et al. 2004; Kemp and Craig 2009; Prematta et al. 2012; Rasmussen et al. 2016; Busse et al. 2021). While erythema marginatum was not identified in any of our patients, formication was identified in some of our patients (46.9%) and pruritus was identified in a few of our patients before the attacks started. These prodromal symptoms were also reported among the possible symptoms (Busse et al. 2021). HAE is a disease with autosomal dominant inheritance pattern. Accordingly, family history also accompanied a great majority of our patients in the study (Busse et al. 2021).
The first angioedema attack can occur at any age, but attacks most frequently start around age 10, and their severity and frequency increase during puberty (Nzeako et al. 2001). However, frequent attacks even in patients as young as 1 to 2 years old, long before puberty, were seen in our patients. This result suggests that this rare disease has not routinely been recognized and clinicians may not diagnose due to this reason. Therefore, there is a need for further studies with pediatric patients (Busse et al. 2021).
HAE might affect many regions in patients. To our knowledge, a great majority of the patients had this disease on their extremities, face, and tongue, while abdominal attacks were seen less frequently (Farkas et al. 2017). In the literature, however, there are also some studies claiming abdominal attacks are observed frequently, contrary to what has been believed (Bork et al. 2006; Sabharwal and Craig 2017; Maurer et al. 2022). We observed the disease on the hands of our patients most frequently (90.6%), but there were abdominal attacks in a considerable number of our patients (71.8%) as well. Since abdominal pain is seen often, especially in children, those who only have abdominal attacks might not be noticed. In our study, a patient was under investigation since he had only frequent abdominal pain. Other causes of abdominal pain were excluded, and the results of his abdominal tomography and endoscopy were found to be normal. Then, C4 and C1-INH were checked to rule out the possibility of HAE. He was diagnosed with HAE, and he responded to treatment with Icatibant (a bradykinin B2 receptor antagonist) and Cinryze (a human plasma-derived C1 esterase inhibitor). Angioedema in another region was not reported in either the history or follow-up of this patient.
It is reported that levels between 81% and 96% of C4 are considered as diagnostic findings for HAE (Yasuno et al. 2021). Similar proportions were observed in our study as well. Nevertheless, the existence of patients whose C4 was normal and whose C1-INH was low required checking if the patient had clinical findings as well. It was observed that C1-INH activity level and genetic examination needed to be checked in some patients to make the diagnosis.
Of all our patients, 93.8% of them had low C4 and 84.4% of them had low C1-INH. C1-INH activity levels were checked in the patients who had normal C1-INH but angioedema clinically; it was to be found low.
Delays in diagnosis have also been reported in other studies, since HAE is a rare disease and it is difficult to diagnose (Frank et al. 1976; Burks et al. 2020). The mean delay was 3 years in our patients. The patients had history of approximately 70 attacks before diagnosis. A 17-year-old female patient who was referred to the Allergy Clinic with suspicion of drug-induced angioedema but was diagnosed with hereditary angioedema was one of the surprising patients. However, she was diagnosed with hereditary angioedema. While the patient’s history was being recorded, she did not report any history of angioedema. When the history of the patient was analyzed thoroughly, it was revealed that she had skin edema due to blunt force, but she did not consult a doctor since those swellings were thought to result from trauma. Based on this patient, we believe that it is necessary to inform the public about this rare disease.
The diagnosis of HAE is often made after recurrent angioedema attacks because it is not routinely diagnosed due to the rareness of the disease. Consequently, we believe that it is important to raise the awareness of emergency physicians and family practitioners about this disease, since they serve as primary health care. The picture can be confused due to the overlap of symptoms with those of the peculiar diseases of childhood.
The most important limitation of our study is that it was conducted in a single center. Multi-center studies with more patients to reveal the clinical course of HAE in children and its characteristics are necessary.
We would like to acknowledge “https://www.makaletercume.com” for their outstanding statistics services that was provided for this manuscript.
The authors declare no conflict of interest.