Abstract
The concept of infection-related glomerulonephritis (IRGN) has been introduced as adults diagnosed with glomerulonephritis often have coexisting active infections. Furthermore, IgA-dominant IRGN is associated with staphylococcal infections in adults with comorbidities, which often progress to end-stage renal disease. Little is known about IgA-dominant IRGN in children, and no consensus for a management strategy of this condition has been reached. We describe the case of a 9-year-old boy with IgA-dominant IRGN that was diagnosed using specific staining for nephritis-associated plasmin receptor (NAPlr)/plasmin activity and galactose-deficient IgA1 (Gd-IgA1), a marker of IgA nephropathy. The patient was successfully treated using a combination of prednisolone, mizoribine (an immunosuppressive drug), and lisinopril (an angiotensin-converting enzyme inhibitor) and three courses of methylprednisolone pulse therapy. The patient was admitted to our hospital with generalized edema, gross hematuria, proteinuria, hypertension, and renal dysfunction. Hypocomplementemia contributed to a diagnosis of IRGN, although the causative organism was unknown. A renal biopsy performed when the patient presented with nephrotic syndrome showed IgA deposition, positive staining for NAPlr, and negative staining for Gd-IgA1, in addition to findings consistent with IRGN, leading to a pathologic diagnosis of IgA-dominant IRGN. The histological staining for NAPlr/plasmin activity and Gd-IgA1, together with clinical symptoms, could be helpful for diagnosing IgA-dominant IRGN. Our findings indicate that otherwise healthy children can also develop IgA-dominant IRGN. Therefore, early diagnosis and aggressive treatment should be considered when IgA-dominant IRGN is suspected to avoid the possibility of incomplete recovery of renal function.
Introduction
Acute glomerulonephritis is often triggered by bacterial or viral infections, and typically presents as acute nephritic syndrome, characterized by oliguria, hematuria, edema, and hypertension (Nasr et al. 2013). In children, most such cases of glomerulonephritis occur following streptococcal upper respiratory tract infection, known as post-streptococcal acute glomerulonephritis (PSAGN) (Nasr et al. 2013). Moreover, the concept of infection-related glomerulonephritis (IRGN) has emerged owing to the prevalence of ongoing active infections at the time of glomerulonephritis diagnosis in adults (Satoskar et al. 2020). Meeting at least three of the following criteria has been proposed as a requirement for a diagnosis of IRGN: 1) Clinical or laboratory evidence of infection preceding or concurrent with glomerulonephritis; 2) reduced serum complement values; 3) endocapillary proliferative and exudative glomerulonephritis; 4) C3-dominant or co-dominant glomerular immunofluorescence staining; and 5) hump-shaped subepithelial deposits on electron microscopy images (Nasr et al. 2011). Positive glomerular staining for nephritis-associated plasmin receptor (NAPlr) and plasmin activity have also been identified as biomarkers of IRGN (Yoshizawa et al. 2022).
The incidence of IRGN associated with Staphylococcus aureus and other bacteria has recently increased among adults, particularly among older persons with comorbidities such as diabetes and malignancies (Nasr et al. 2013; Satoskar et al. 2020). The IRGN among such patients is characterized by dominant IgA immunoglobulin deposits within glomeruli. These variants of IRGN are termed IgA-dominant IRGN (Nasr et al. 2003). The renal prognosis for adult patients with IgA-dominant IRGN is poor, with a complete recovery rate of only 16% (Haas et al. 2008). In addition, previous reports have shown that 43% of adult patients with IgA-dominant IRGN had persistent renal dysfunction and 41% progressed to end-stage renal disease (ESRD) (Haas et al. 2008). Although IgA-dominant IRGN has been reported to occur in healthy children and to be associated with sequelae of renal function (Rus et al. 2015; Mascarenhas et al. 2016; Srinivasaraghavan et al. 2016; Shirai et al. 2020; Takase et al. 2020; Saghar et al. 2022), robust diagnostic and therapeutic strategies for IgA-dominant IRGN have not been established.
We describe the case of a boy with IgA-dominant IRGN diagnosed using specific staining for NAPlr and galactose-deficient IgA1 (Gd-IgA1). Aggressive immunosuppressive therapy led to complete remission.
Case Presentation
A 9-year-old boy presented to our hospital with generalized edema and gross hematuria. His medical history was unremarkable. At the first visit to our hospital, the patient exhibited hypocomplementemia (C3 8.2 mg/dL, reference value 86-160 mg/dL; C4 28 mg/dL, reference value 17-45 mg/dL; CH50 10 U/mL, reference value 25-48 U/mL), hematuria (3+), and proteinuria [2+, urine protein-to-creatinine ratio (UPCR) 1.2 g/gCr]. Blood pressure and renal function were within normal ranges. The patient was febrile 4 weeks before the medical examination. Based on these observations, the patient was diagnosed with IRGN, and antibiotic treatment with cefaclor (50 mg/kg/day) was initiated and followed up on an outpatient basis. However, one week later, the patient was admitted to the hospital for acute nephritic syndrome with oliguria (urine volume 300 mL/day), worsening edema, renal dysfunction [creatinine-estimated glomerular filtration rate (Cr-eGFR) 78.0 mL/min/1.73 m2] and stage 2 hypertension (blood pressure 135/102 mmHg).
His temperature at admission was 36.5℃, blood pressure was 139/100 mmHg, pulse 20 bpm, and respiratory rate of 20 breaths/min with an O2 saturation of 98% on room air. Gastrointestinal and respiratory symptoms were absent. The periorbital region was edematous as were lower extremities, but purpura was absent on the limbs. The patient weighed 3 kg more from the date of his first visit to our hospital. Table 1 shows the laboratory findings. Urinalysis revealed specific gravity of 1.034, high proteinuria (3+, UPCR 13.8 g/gCr), and hematuria (occult blood, 4+; red blood cells, 20-29/high power field). Renal dysfunction was evident, with serum creatinine at 0.56 mg/dL (Cr-eGFR 78.0 mL/min/1.73 m2), which was a significant increase from 0.40 mg/dL (Cr-eGFR 108.8 mL/min/1.73 m2) at the initial presentation. Immunological findings were negative, except for a substantially decreased complement fraction C3 (7.4 mg/dL). The C4 level was normal (19.1 mg/dL) and levels of the antibodies anti-streptolysin-O (ASO) and anti-streptokinase (ASK) were not elevated. Although the causative organism was not identified, IRGN was suspected based on clinical symptoms and blood and urine test results, as at the initial visit.
The patient was treated with cefaclor (30 mg/kg/day), 2.5 mg oral amlodipine once a day, furosemide (1 mg/kg/dose), and dietary salt restriction from the time of admission, which improved the hypertension and renal dysfunction. Serum C3 levels were completely normalized by day 9 after admission. However, proteinuria (UPCR approximately 20 g/gCr) persisted and progressed to nephrotic syndrome. Therefore, a percutaneous ultrasound-guided kidney biopsy was obtained on day 10 after admission.
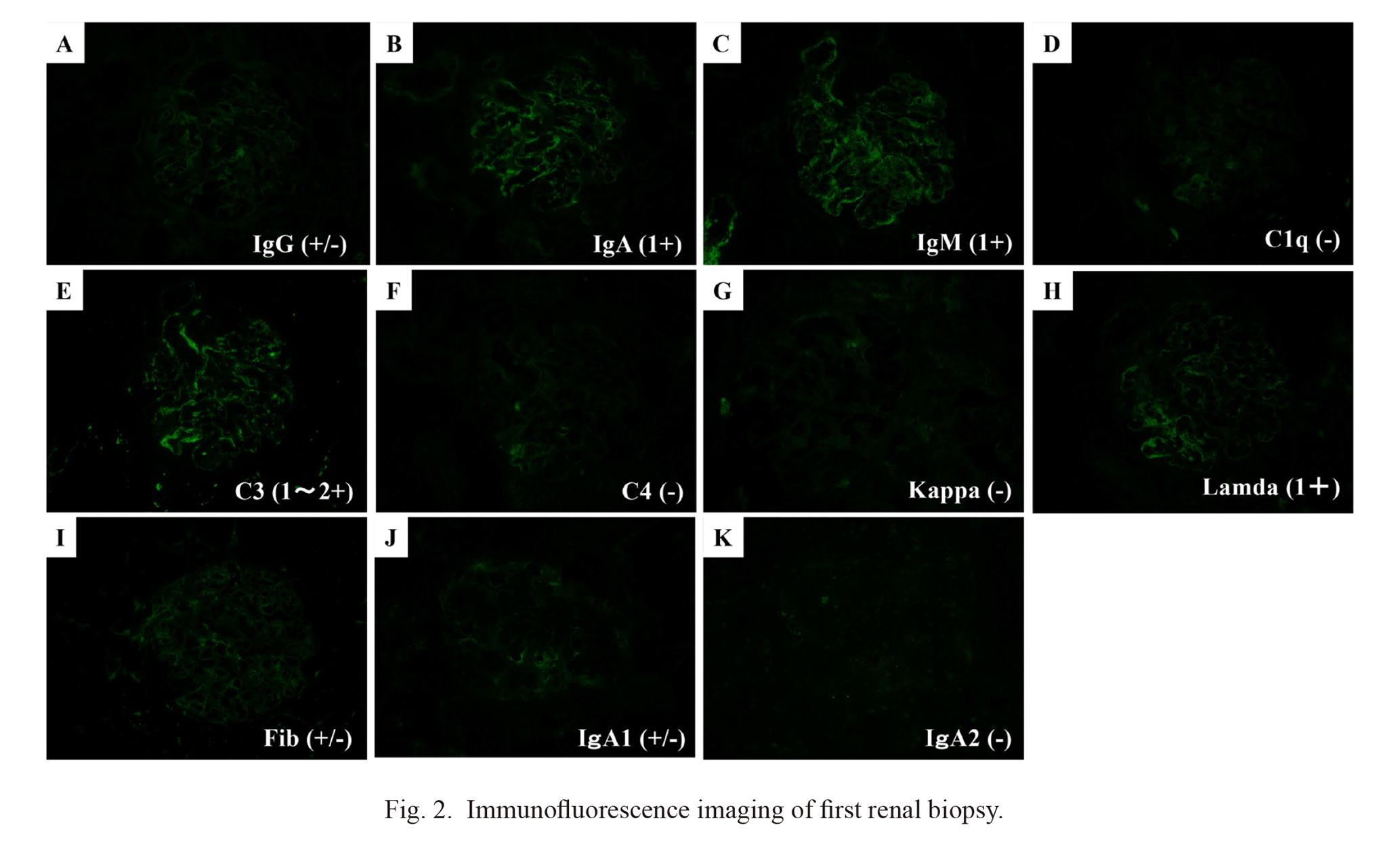
Light microscopy revealed 36 glomeruli with diffuse global endocapillary proliferative glomerulonephritis, accompanied by neutrophil and macrophage exudation (Fig. 1A). Three of these glomeruli contained cellular crescents (Fig. 1B). No adhesions, segmental glomerulosclerosis, or tubulointerstitial changes were evident. Electron microscopy findings revealed hump-shaped, subepithelial electron-dense deposits (Fig. 1C). Immunofluorescence findings (Fig. 2) were positive for IgA (1+; Fig. 2B) and C3 (1-2+; Fig. 2E) in mesangial and capillary areas. Immunofluorescence staining of IgA subclasses revealed slightly predominant IgA1 deposition (Fig. 2J, K). Furthermore, immunofluorescence staining for NAPlr and in situ zymography for plasmin activity, biomarkers of IRGN, were both positive in the segmental areas of the glomeruli in similar distribution (Fig. 3A, B). On the other hand, immunohistochemical staining for Gd-IgA1 in glomeruli using an anti-Gd-IgA1 monoclonal antibody (KM55) was negative (Fig. 3C). A final diagnosis of IgA-dominant IRGN in this patient was confirmed by the pathological findings of the biopsy.
Prednisolone (60 mg/m2/day) was started on hospitalization day 21, considering nephrotic syndrome (Fig. 4). However, despite prednisolone treatment for 2 weeks, proteinuria persisted (UPCR > 2.0 g/gCr). Therefore, the therapeutic regimen for severe childhood IgA nephropathy (IgAN) in Japan was applied considering the IgA deposition (Yoshikawa et al. 2008).
Three courses of intravenous methylprednisolone pulse therapy (MPT, 30 mg/kg/dose × 3 days) were administered, followed by a combination of prednisolone (2 mg/kg, administered on alternate days), lisinopril (an angiotensin-converting enzyme inhibitor, 0.4 mg/kg/day), and mizoribine (an immunosuppressive drug, 4 mg/kg/day) (Fig. 4). The multi-drug combination resulted in negative nephrotic proteinuria at 123 days after onset. Serum creatinine levels improved to 0.27 mg/dL (Cr-eGFR; 158.6 mL/min/1.73 m2). Prednisolone was tapered by 0.5 mg/kg at 4-week intervals to 25 mg (1 mg/kg) over 3 months.
A second renal biopsy approximately 2 months after the initial biopsy was required due to another episode of worsening renal function that occurred during the disease course. Although the endocapillary hypercellularity was improved, mesangial cell proliferation was prominent. There was no crescentic formation in the obtained 55 glomeruli. These findings suggested that the specimen showed recovery and chronic phases of IRGN (Fig. 5A, B). In addition, electron microscopy suggested a washing-out appearance of the electron-dense deposits (Fig. 5C). Immunofluorescence staining showed IgA1-dominant deposition in IgA subclasses, and IgA1 positivity was clearer than that of the initial biopsy (Fig. 5D, E). The glomerular deposition of C3 was not apparent (Fig. 5F).
The patient continues to receive multi-drug combination therapy (prednisolone, 1 mg/kg every alternate day). At approximately 1 year after discharge from the hospital, neither proteinuria nor renal dysfunction recurred, and only microscopic hematuria persists.
Our hospital Ethics Committee does not require informed consent for a single patient case report.
Discussion
We diagnosed pediatric IgA-dominant IRGN based on pathological NAPlr and Gd-IgA1 staining. This is the first report of a child who achieved complete remission of IgA-dominant IRGN after treatment with a combination of prednisolone, mizoribine, and lisinopril. Although IgA-dominant IRGN can lead to ESRD, pediatric data on this is limited. Thus, whether the renal prognosis is more favorable among children than in adults and older populations remains unknown. In addition, diagnostic methods and treatment for IgA-dominant IRGN have not yet been established. Table 2 provides a summary of publications describing IgA-dominant IRGN in children. Among children with IgA-dominant IRGN, to the best of our knowledge, our patient is the only one diagnosed based on NAPlr and Gd-IgA1 staining. Reports of pediatric IgA-dominant IRGN vary in terms of confirmation of infection cause, treatment modalities, and outcomes. Some patients have experienced complete improvement in renal function with angiotensin-converting enzyme inhibitors alone, whereas others, including children without underlying medical conditions, have progressed to ESRD despite aggressive MPT and blood purification.
The diagnosis of IRGN in our patient was not difficult because all diagnostic criteria for IRGN (Nasr et al. 2011) were met, and positive staining for NAPlr and plasmin activity was similarly distributed in glomeruli. Our patient had non-group A Streptococcus-induced IRGN, which could explain the weak staining for NAPlr/plasmin activity (Uchida and Oda 2020).
Serum Gd-IgA1 is one of the most important biomarkers for diagnosing IgAN (Yasutake et al. 2015). Furthermore, Gd-IgA1 immunofluorescence staining on renal biopsy specimens is specifically found in the glomeruli of patients with IgAN and IgA vasculitis and aids in diagnosis (Suzuki et al. 2018). The biopsy specimens from our patient stained negative for Gd-IgA1, which differed from the findings of patients with IgAN. The intensity of Gd-IgA1 staining is significantly lower in patients with IgA-dominant IRGN than in those with primary IgAN, and as such, Gd-IgA1 staining intensity might help distinguish between them (Zhang et al. 2021). In contrast, IgA-dominant IRGN showing high titer of serum Gd-IgA1 and positive staining of renal tissue for Gd-IgA1 has also been reported (Han et al. 2021). Gd-IgA1 is reported to be overproduced by interleukin-6 during infection (Suzuki et al. 2014), and IgA-dominant IRGN cannot be excluded when patients are Gd-IgA1-positive. Therefore, this staining alone cannot be used as a specific diagnostic method for IgA-dominant IRGN. However, characteristic clinical features such as acute kidney injury (AKI), nephrotic proteinuria, and hypocomplementemia support a diagnosis of IgA-dominant IRGN (Haas et al. 2008). Therefore, we believe that IgA-dominant IRGN can be diagnosed by combining clinical symptoms with specific staining, such as Gd-IgA1. We diagnosed our patient with IgA-dominant IRGN based on previous infections, clinical manifestations of AKI, hematuria, nephrotic-range proteinuria, and hypocomplementemia together with the findings of specific immunohistochemical staining.
Antibiotics have been the cornerstone of treatment for IRGN, but immune suppression data for IRGN is limited. However, the combination of prednisolone and aggressive immunosuppressive therapy has the potential to improve IgA-dominant IRGN (Kikuchi et al. 2006; Okumura et al. 2022).
One limitation of this study is that we cannot ascertain whether the patient would have also achieved remission without multi-drug combination therapy.
The distinct prognosis and therapeutic considerations for IgA-dominant IRGN, IgAN, and PSAGN highlight the need for increased awareness of IgA-dominant IRGN among pediatricians. Considering the potential for incomplete renal recovery and progression to ESRD, extensive research is warranted to establish specific diagnostic methods and strategies for treating IgA-dominant IRGN.
In conclusion, we diagnosed pediatric IgA-dominant IRGN using NAPlr and Gd-IgA1 staining. The patient achieved complete remission due to aggressive immunosuppressant therapy using steroids and immunosuppressant drugs. These results highlight the importance of early diagnosis and intervention. Future research efforts should be undertaken to develop robust management strategies and could incorporate our approach.
Author Contributions
Dr. Takagi collected the information, and drafted and revised the manuscript. Dr. Kano, Dr. Oda, Dr. Suzuki, Dr. Ono, and Prof. Yoshihara interpreted all data and critically revised the manuscript for important intellectual content. All authors have approved the final manuscript and agreed to be accountable for all aspects of this work.
Conflict of Interest
The authors declare no conflict of interest.
References
-
Haas,
M.,
Racusen,
L.C. &
Bagnasco,
S.M.
(2008) IgA-dominant postinfectious glomerulonephritis: a report of 13 cases with common ultrastructural features. Hum. Pathol., 39, 1309-1316.
-
Han,
W.,
Suzuki,
T.,
Watanabe,
S.,
Nakata,
M.,
Ichikawa,
D.,
Koike,
J.,
Oda,
T.,
Suzuki,
H.,
Suzuki,
Y. &
Shibagaki,
Y.
(2021) Galactose-deficient IgA1 and nephritis-associated plasmin receptors as markers for IgA-dominant infection-related glomerulonephritis: a case report. Medicine (Baltimore), 100, e24460.
-
Kikuchi,
Y.,
Yoshizawa,
N.,
Oda,
T.,
Imakiire,
T.,
Suzuki,
S. &
Miura,
S.
(2006) Streptococcal origin of a case of Henoch-Schoenlein purpura nephritis. Clin. Nephrol., 65, 124-128.
-
Mascarenhas,
R.,
Fogo,
A.B.,
Steele,
R.W. &
Baliga,
R.
(2016) IgA-Dominant postinfectious glomerulonephritis. Clin. Pediatr. (Phila.), 55, 873-876.
-
Nasr,
S.H.,
Fidler,
M.E.,
Valeri,
A.M.,
Cornell,
L.D.,
Sethi,
S.,
Zoller,
A.,
Stokes,
M.B.,
Markowitz,
G.S. &
D’Agati,
V.D.
(2011) Postinfectious glomerulonephritis in the elderly. J. Am. Soc. Nephrol., 22, 187-195.
-
Nasr,
S.H.,
Markowitz,
G.S.,
Whelan,
J.D.,
Albanese,
J.J.,
Rosen,
R.M.,
Fein,
D.A.,
Kim,
S.S. &
D’Agati,
V.D.
(2003) IgA-dominant acute poststaphylococcal glomerulonephritis complicating diabetic nephropathy. Hum. Pathol., 34, 1235-1241.
-
Nasr,
S.H.,
Radhakrishnan,
J. &
D’Agati,
V.D.
(2013) Bacterial infection-related glomerulonephritis in adults. Kidney Int., 83, 792-803.
-
Okumura,
M.,
Sugihara,
S.,
Seki,
K.,
Nagaoka,
K.,
Okawa,
N.,
Ebihara,
M.,
Inoue,
T.,
Fukuda,
J.,
Ohara,
M.,
Imasawa,
T.,
Kitamura,
H.,
Oda,
T. &
Suzuki,
T.
(2022) Use of immunosuppressive therapy in the treatment of IgA-dominant infection-related glomerulonephritis. Intern. Med., 61, 697-701.
-
Rus,
R.R.,
Toplak,
N.,
Vizjak,
A.,
Mraz,
J. &
Ferluga,
D.
(2015) IgA-dominant acute poststreptococcal glomerulonephritis with concomitant rheumatic fever successfully treated with steroids: a case report. Croat. Med. J., 56, 567-572.
-
Saghar,
A.,
Klaus,
G.,
Trutnau,
B.,
Kömhoff,
M.,
Gröne,
H.J. &
Weber,
S.
(2022) A rare case of Immunoglobulin A dominant post-infectious glomerulonephritis (IgA PIGN) in a young patient. BMC Nephrol., 23, 333.
-
Satoskar,
A.A.,
Parikh,
S.V. &
Nadasdy,
T.
(2020) Epidemiology, pathogenesis, treatment and outcomes of infection-associated glomerulonephritis. Nat. Rev. Nephrol., 16, 32-50.
-
Shirai,
Y.,
Miura,
K.,
Yabuuchi,
T.,
Nagasawa,
T.,
Ishizuka,
K.,
Takahashi,
K.,
Taneda,
S.,
Honda,
K.,
Yamaguchi,
Y.,
Suzuki,
H.,
Suzuki,
Y. &
Hattori,
M.
(2020) Rapid progression to end-stage renal disease in a child with IgA-dominant infection-related glomerulonephritis associated with parvovirus B19. CEN Case Rep., 9, 423-430.
-
Srinivasaraghavan,
R.,
Krishnamurthy,
S.,
Dubey,
A.K.,
Parameswaran,
S.,
Biswal,
N. &
Srinivas,
B.H.
(2016) IgA dominant post-infectious glomerulonephritis in a 12-year-old child. Indian J. Pediatr., 83, 470-472.
-
Suzuki,
H.,
Raska,
M.,
Yamada,
K.,
Moldoveanu,
Z.,
Julian,
B.A.,
Wyatt,
R.J.,
Tomino,
Y.,
Gharavi,
A.G. &
Novak,
J.
(2014) Cytokines alter IgA1 O-glycosylation by dysregulating C1GalT1 and ST6GalNAc-II enzymes. J. Biol. Chem., 289, 5330-5339.
-
Suzuki,
H.,
Yasutake,
J.,
Makita,
Y.,
Tanbo,
Y.,
Yamasaki,
K.,
Sofue,
T.,
Kano,
T. &
Suzuki,
Y.
(2018) IgA nephropathy and IgA vasculitis with nephritis have a shared feature involving galactose-deficient IgA1-oriented pathogenesis. Kidney Int., 93, 700-705.
-
Takase,
Y.,
Nakayama,
Y.,
Urakawa,
T.,
Akaishi,
S.,
Yamada,
H.,
Sakamoto,
A.,
Shirakawa,
T.,
Nakashima,
Y.,
Moriuchi,
H. &
Nakashima,
K.
(2020) Childhood acute nephritic syndrome with histopathological features of IgA-dominant infection-related glomerulonephritis: a case report. J. Jpn. Pediatr. Soc., 124, 1733-1739.
-
Uchida,
T. &
Oda,
T.
(2020) Glomerular deposition of nephritis-associated plasmin receptor (NAPlr) and related plasmin activity: key diagnostic biomarkers of bacterial infection-related glomerulonephritis. Int. J. Mol. Sci., 21, 2595.
-
Yasutake,
J.,
Suzuki,
Y.,
Suzuki,
H.,
Hiura,
N.,
Yanagawa,
H.,
Makita,
Y.,
Kaneko,
E. &
Tomino,
Y.
(2015) Novel lectin-independent approach to detect galactose-deficient IgA1 in IgA nephropathy. Nephrol. Dial. Transplant., 30, 1315-1321.
-
Yoshikawa,
N.,
Nakanishi,
K.,
Ishikura,
K.,
Hataya,
H.,
Iijima,
K. &
Honda,
M.;
Japanese Pediatric IgA Nephropathy Treatment Study Group
(2008) Combination therapy with mizoribine for severe childhood IgA nephropathy: a pilot study. Pediatr. Nephrol., 23, 757-763.
-
Yoshizawa,
N.,
Yamada,
M.,
Fujino,
M. &
Oda,
T.
(2022) Nephritis-associated plasmin receptor (NAPlr): an essential inducer of C3-dominant glomerular injury and a potential key diagnostic biomarker of infection-related glomerulonephritis (IRGN). Int. J. Mol. Sci., 23, 757-763.
-
Zhang,
M.,
Zhou,
W.,
Liu,
S.,
Zhang,
L.,
Ni,
Z. &
Hao,
C.
(2021) KM55 monoclonal antibody staining in IgA-dominant infection-related glomerulonephritis. Nephron, 145, 225-237.