Reviews for award
Discovery of Bonding-active Chemical Species Containing Nitrogen Atoms
2023 Volume 143 Issue 4 Pages 323-336
Details
2023 Volume 143 Issue 4 Pages 323-336
In this review, the authors review and explain their research on “Discovery of Bonding Active Species Containing Nitrogen Atoms” from the past to the present. The authors are interested in new chemical phenomena, especially in the activation of chemical bonds containing nitrogen atoms, and have conducted research to discover chemical bonds with new properties. The activated chemical bonds containing nitrogen atoms are the following (Fig. 1). (1) Rotationally activated C–N bonds by pyramidalization of amide nitrogen atoms (2) N–N bond cleavage ability with reduced bond strength by pyramidalization of nitrosamine nitrogen atoms (3) Transient hetero atom-N bond formation by neighboring group participation of a halogen electron to the nitrogen cation. (4) A unique carbon cation reaction involving nitrogen atoms, especially nitro groups (C–NO2 bond) and ammonium ions (C–NH3+ bond). These purely basic chemistry discoveries unexpectedly led to the creation of functional materials, especially biologically active molecules. We will explain how new chemical bonds led to the creation of new functions.
本総説では筆者らが過去から現在までに行った「窒素原子を含む結合活性化学種の発見」に関する研究を概観し解説する.
筆者らは新しい化学現象,特に窒素原子を含む化学結合の活性化に興味を持ち,新たな性質を持つ化学結合を発見する研究を行ってきた.窒素原子を含む活性化された化学結合とは以下のものである(Fig. 1).(1)アミド窒素原子の非平面化による回転活性されたC–N結合の研究,(2)ニトロソアミン窒素原子の非平面化による結合強度の減少したN–N結合の開裂能の研究,(3)窒素カチオンへのハロゲン電子の隣接基関与による一過的なN–ハロゲン結合の形成,(4)窒素原子,特にニトロ基(C–NO2結合)やアンモニウムイオン(C–NH3+結合)が関与する特異な炭素カチオン反応,である.

これらの純粋に基礎化学の発見が,思いがけず機能ある物質,特に生物活性のある分子の創製につながることを示した(Fig. 2).新しい化学結合が新しい機能創出につながったことを解説する.
この研究の発端はアミド結合の平面性にある.アミド結合はFig. 3の共鳴構造が書けることからアミドのC–N結合は二重結合性を持ち平面構造を取る.一方で,もしアミド窒素が平面(sp2混成)ではなくピラミッド化(sp3混成)すると,共鳴による安定化が失われ,出現する非平面アミドは不安定で,水で容易に加水分解されるため構造単位としては利用できない.1)ところが偶然にも,新たな安定な窒素ピラミッドアミドに遭遇した.このアミド結合をペプチドとして利用し,そのホモオリゴマーを合成するとヘリックス構造を取ることがわかった.このヘリックスはタンパク質–タンパク質相互作用を阻害した.なお,この非平面アミドの初期研究について2001年に薬学雑誌に総説を発表している.2)また最近成書にも発表している.3)

前述のように,アミド結合は一般的には平面構造を取るが,二環性構造である7-アザビシクロ[2.2.1]ヘプタン[7-azabicyclo[2.2.1]heptane(Abhと記述することがある)]に組み込むと,アミド結合の窒素がピラミッド化することを発見した(Fig. 4).4)この7-azabicyclo[2.2.1]heptane骨格はπ面選択の研究テーマの中で扱っていた(未発表).5) Figure 5のアミン窒素は反転が遅いことが知られているが(後述),6)反転は平均化されるとみなすと,あたかも平面分子と近似でき,窒素原子の被占2p軌道に対する面選択性が議論できる.求電子的な酸化反応(N-オキサイド化)を検討した.

The activation energy for the nitrogen inversion is small and freely reversible.

片方のエタノブリッジにはCF3基が2つ下向きに置換しているN-ethyl-7-azabicyclo[2.2.1]heptaneを合成した.求電子的な酸化反応(N-オキサイド化)は大きな面選択性を示した.アミンのピラミッド化の平衡はNMRで59 : 41の多少の偏りが認められたがこの程度を上回る面選択がある(Fig. 5).
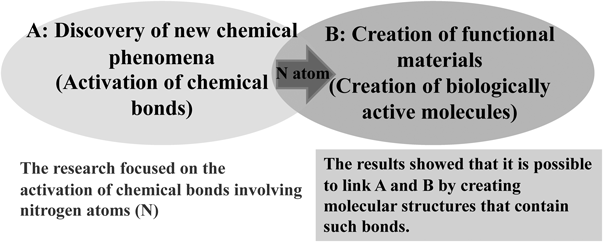
このN-ethyl-7-azabicyclo[2.2.1]heptaneの原料はN-アセチル体であるアミドである.筆者はなぜか対応するアミドは“twisted amide”ではないだろうかと思い込みX線結晶解析を山口健太郎博士にお願いしたところ,窒素ピラミッド化していることを発見した.2,3)世の巡り合わせとは妙なもので,その時期に,7-azabicyclo[2.2.1]heptaneのアミン窒素は反転が遅いことを発表していたStephen F. Nelsen教授が偶然にも首藤紘一先生を訪ねてきて研究室内で小さなセミナーを行って頂いた.セミナーの後,首藤先生のお供でNelsen先生と食事に行ったが,アミドの非平面化の話はしなかった.既にお気づきだと思うが,Nelsen教授はアミンの反転減速をBicyclic Effectと名付けられて論文を発表していたが,6)アミドのピラミッド化が同じような構造要因である可能性示唆している.つまり窒素周りの平面構造が不安定であることを示唆している.窒素ピラミッドアミドのピラミッド化の方向にアミンと同様に左・右の2通りあり,早い平衡にある(Fig. 6).β-プロリンを2つの炭素原子で架橋したこの構造を二環性βプロリンとも呼ぶことにする.この非天然アミノ酸の作るアミド結合は,二重結合性が減弱して回転し易くなっている.7) Figure 6に示したアミドではcis体とtrans体は等価であるが(ピラミッド化による窒素原子のキラリティーは考えないとする)後に出てくるカルボン酸をβ位に置換したβ-アミノ酸誘導体の場合は,アミノ酸の主鎖に沿って本総説ではアミドのcis/transを定義する(一般的なE/Zの定義と異なる場合がしばしばあるが,アミノ酸の世界のなかでは統一が保たれている).通常非平面アミドは加水分解されやすく不安定な化合物であり,ピラミッドアミドを作る小員環アミン(アジリジンやアゼチジン)のアミドは不安定ではないものの化学的な加工や後で話題になるアミドの回転異性体の制御は容易ではない.1)さらに,筆者らが見つけた窒素ピラミッドアミドはむしろアルカリ加水分解に抵抗するし,8)酸性でも安定であった.
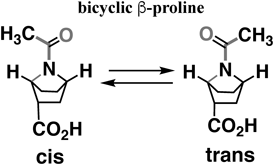
この二環性βプロリンアミドには赤色で示したペプチドの主鎖に沿ってシス体とトランス体の2つの異性体があり,この場合には,50 : 50の混ざりとして存在している.9) β-アミノ酸からなるペプチドは,α-アミノ酸からなるペプチドに比べ炭素結合の回転自由度が増え規則構造を取り難いと考えられるが,Seebach10)やGellman11)が示したように,β-アミノ酸からなるペプチドは,α-アミノ酸からなるペプチド(12–16残基)よりはるかに短い6残基程度でヘリックス構造を誘起することが示された.この二環性βプロリンを一般的なβ-アミノ酸及びペプチドと構造の自由度を比較してみると,β-アミノ酸ペプチドにおいて,NHアミドを形成するとアミド結合は動的にトランスに固定されているので(ω=180.0°),結合の回転の自由度は,ϕ, θ, ψと自由度は3と考えられる(Fig. 7, left).一方,二環性βプロリンアミドでは,二環性構造ゆえに自由度が少ない分子にみえるが,ピラミッド化に由来する角度α,アミドの早い回転に由来する角度ω,一重結合の回転ψの3つの自由度がある(Fig. 7, right),一般的なβ-アミノ酸ペプチド(Fig. 7, left)と同じ数の自由度を持っている.アミド窒素ピラミッド化によって二環性βプロリンアミドはフレキシブルな分子とみなすことができ,α-アミノ酸より短鎖でヘリックス構造を形成することから,9)自由度の高さは,動的規則構造化にとって重要な因子であると考えられる.

アミドのシス–トランス平衡をどちらかに片寄らせることを考えた.橋頭位4位に置換基を導入するとRとアミドの主鎖の立体反発でシス体が100%になることが判明した(Fig. 8).12)またもう1つのブリッジヘッドである1位に置換基を導入すると立体反発で,今度はトランス体が100%になった.13)この現象は事前にDFT計算でも予想されていた.

このシスアミドやトランスアミドのユニットを光学分割で(S)体,(R)体の両方を合成し,(S)体,(R)体のホモオリゴマー,単量体,2量体,4量体,6量体,8量体をそれぞれ合成した.Circular dichroism(CD)スペクトルで,鎖長依存性,溶媒効果,濃度効果,温度効果を調べたが,1残基当たりのCD強度は一定で,1残基で既に規則構造を取り始めている.12,13)シス体では二量体のX線結晶解析が,トランス体では三量体のX線結晶解析に成功したので,8量体の構造まで延長するとFig. 9に示すように,ホモオリゴマーはヘリックス構造を取ることがわかった.窒素上に水素がないことから,水素結合によらない,つまり水中でも安定なヘリック構造が創製できた(Fig. 9).シスアミドのヘリックスでは4残基で1回転し,トランスアミドのヘリックスでは2.7残基で1回転する.天然α-アミノ酸の作るαヘリックスが3.6残基で1回転するのとは異なり,ピッチの長さや直径も異なる規則性を持っている.一回転で進むピッチ長が天然アミノ酸のαヘリックスより長い.

このヘリックスは,αヘリックスの相互作用によって結合するPPIを阻害することがわかった.14,15)
この化合物はP53の代わりにMDM2に結合した(Fig. 10).MDM2の広い相互作用面を3つのユニットでカバーできる.

Three bold amino acid side chains correspond respective to the key reside of p53 which interact with the MDM2 Helix.
ラクタムアミドのアミド結合は普通360度回転しようとすると環が巻き付いて回転できない.この現象は環のサイズによらない.360度回転し続けることはできない(Fig. 11).

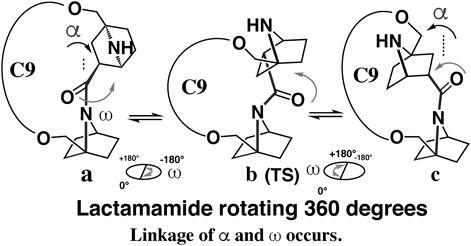
ところが,この二環性βプロリンのダイマーを含むラクタムでは二面角αとωが連動しラクタムアミド結合が360度回転できることがわかった(Fig. 12).16)ラクタム環の大きさとアミドの回転速度におおよその直線関係があることがわかった.分子動力学計算による溶媒和を含んだギブス自由エネルギー変化からアミド回転は連続的で,アミド酸素が手前から奧に回転するとき,アミドは右側に倒れ,奧から手間に回転するときアミドは左側に倒れることによって360度回転できることが推定された.アミド窒素ピラミッド化は重要な自由度の1つであることを示している.
ところで,βストランド構造は通常βシート構造にみられる.すなわちβストランド間の水素結合で安定化される構造で,1本鎖のアミノ酸のβストランド構造はあまり研究されていない(Fig. 13, center).筆者らが見い出した二環性βプロリンをαアミノ酸配列に組み込んだヘテロオリゴマーでは,N末,C末のアミノ酸にβストランド構造を誘起することが明らかになった(Fig. 13, right).17) βストランド構造で有名なAβ40(Fig. 13, left)の疎水性アミノ酸残基間の電子相互作用の考察も行った.疎水性相互作用は原子間相互作用の可能性がある.18)

ところで二環性構造で,窒素ピラミッド化を起こす系は7-azabicyclo[2.2.1]heptane構造だけであろうか.この問いは当初からあるもので,最近分子動力学計算とdensity functional theory(DFT)計算を組み合わせることで評価を実施し,7-azabicyclo[2.2.1]heptane以外の構造でも窒素ピラミッドアミドが形成できる可能性を報告した.19)
N-ニトロソアミンはアミドと類似の共鳴構造が書けるのでN=N二重結合性を持ち通常は平面構造を取る(Fig. 14).ニトロソアミンの窒素原子を7-azabicyclo[2.2.1]heptaneに組み込むこと,窒素がピラミッド化したニトロソアミンを安定に合成できることがわかった(Fig. 15).20,21)


ニトロソアミンの窒素原子がピラミッド化によって二重結合性が弱くなったN–NO結合のおかげでシステインなどのチオールと反応して,S-トランスニトロソ化反応を起こした(Fig. 15).22) S-ニトロソ化[生物論文ではS-ニトロシル化(S-nitrosylation)と呼ばれる]はタンパク質翻訳後修飾の1つで,情報伝達においてリン酸化に匹敵する重要性を持っていると言われている.23)窒素ピラミッドニトロソアミンによるS-トランスニトロソ化がG-タンパク質共役型受容体(G protein-coupled receptor: GPCR)の脱感作の抑制やtransient receptor potential ankyrin(TRPA)チャネルの選択的開口への応用につながった(Fig. 16).

これらの生物現象はタンパク質の特定のシステインのS-ニトロソ化によって引き起こされる.S-ニトロソ化がGPCRをリン酸化するキナーゼであるGRK2のシステインで起きると,アゴニスト(イソプレテノール)刺激におけるβ2アドレナリン受容体の脱感作(細胞膜表面からの受容体の引き込み)を抑制することがわかった[Fig. 16(a)].24)またこのニトロソアミンが,数あるTRPチャネルファミリーのうちTRPAチャネルを選択的に開口した[Fig. 16(b)].25) TRPAチャネルは複数のシステインのS-ニトロソ化により開口することが知られている.
3-1. ニトロソアミンのN–NO結合の可視光照射による結合開裂.NO-Cage化合物ところで窒素ピラミッド化したニトロソアミンのn→π*遷移に由来するUV-可視吸収スペクトルは,窒素平面ニトロソアミンよりも長波長に吸収を持つ(Fig. 17).

これはFig. 18に示す軌道相互作用におけるhighest occupied molecular orbital(HOMO)–lowest unoccupied molecular orbital(LUMO)間の軌道エネルギーの差の減少によるものである.可視光に吸収ピークがあると言うことは,可視光を照射すると可視光を吸収して電子のHOMO→LUMO励起が起こる.LUMOはN–NO結合の反結合性のπ*軌道であるため,HOMOから電子が励起してLUMOに入るとN–NO結合がラジカル的に開裂して,·NO(一酸化窒素)と·N<が生成する(Fig. 19).26)一酸化窒素検出蛍光プローブとして知られるdiaminofluorescein-2(DAF2)は,実際は·NOのトラップ剤ではなく,溶存酸素で·NOがNO2−に酸化され,生成する+NOがDAF2と反応して蛍光を発する.可視光照射の溶媒を脱気して溶存酸素を取り除くと蛍光発色が起きないことから,可視光照射で生成するのはN–NO結合のホモ開裂で生成する·NOであることがわかる.


隣接基関与は炭素カチオンについて多く研究されているが,他のカチオンへの隣接基関与の例はあまり目にしない.筆者らは,sp2型の窒素カチオンが隣接するBrによって一過的に結合が形成され新たな反応性を示すことを明らかにした(Fig. 20).窒素カチオンへの隣接基関与の最初の例である.

ブレステッド酸を用いるオキシムのベックマン転位反応を検討したときに遭遇した予想外の化合物の生成がきっかけである.
筆者らは細胞膜に存在するある種のカリウムイオンチャネル(BKチャネル)の開口を引き起こす生物活性物質を研究していて,27)天然物のデヒドロアビエチン酸の骨格を持つ誘導体を多数合成していた.誘導化の1つとして,B環のラクタム化を考えた[Fig. 20(a)].
トリフルオロ酢酸(trifluoroacetic acid: TFA)を用いるベックマン転位反応条件下で,デヒドロアビエチン酸メチルエステルの7位ケトオキシム誘導体(実際は酸素–トシル体)から生成する化合物を検討した.28)その際,偶然にも,そしてその後何年にもわたって頭を悩まし続けることになる予想外の反応に遭遇した.12位にクロロ基を有する場合,通常のベックマン転位が進行し,ベンゼン側のC–C結合が転位した(以下,ベンゼン転位(=ベックマン転位)と呼ぶ)ラクタムが単一生成物として得られた.一方,同じクロロ基がペリ位に当たる14位に存在すると,同一の反応条件下で,2種類のラクタムが生成した.生成比に大きな違いがあるため,ほとんど1種類のラクタムが主生成物と言ってよい.驚いたことに,その主生成物は当然生成すると信じていたベンゼン側のC–C結合が転位したラクタムではなく,アルキル側のC–C結合が転位した(以下,アルキル転位と呼ぶ)ラクタムであることが判明した.わずかに副生するもう1つの生成物が,通常のベックマン生成物であるベンゼン側のC–C結合が転位したラクタムであった.この全く予想外の反応生成物を得たため,その一般性を類似の14位臭素置換体でも調べたところ,完全な選択性を持ってアルキル側のC–C結合が転位したラクタムを単一の生成物として与えた[Fig. 20(a)].すべてのオキシムにおいてオキシムのN–O結合はベンゼン環に対してアンチであることを,合成したすべてのオキシムのX線結晶構造解析を行うことで確認・検証した.また,オキシムのN–O結合のシン体への異性化はTFA存在下の反応条件下はもちろん,TFA中加熱しても起きないことを確認しているので,アルキル転位体の生成はオキシムの異性化のせいではないことは明らかだった.
近傍に位置する窒素カチオンの間に新しいハロゲン–窒素結合形成反応機構の解明と反応性の一般性を調査するため,デヒドロアビエチン酸メチルエステルの部分構造である1-テトラロンオキシム誘導体の転位生成物を調べた[Fig. 20(b)].TFA中同様な酸素–トシルオキシムの窒素原子のペリ位に当たる芳香環上にハロゲン原子[塩素(Cl),臭素(Br)]が存在すると,この窒素上の酸素–トシル基(OTs基)と同じ側にあるアルキル側のC–C結合が優先的に転位するアルキル転位が起きることがわかった[Fig. 20(b)].一方,オキシム窒素の近傍に水素(H)あるいはハロゲン原子でもフッ素(F)が存在するとベンゼン側の炭素–炭素結合が転位する従来の「ベックマン転位」が優先的にみられた.27)ペリ位に求核的なハロゲン原子が存在すると,「ベックマン転位」の反応条件では通常はみられないアルキル転位反応が進行し,ベックマン転位とは異なる「新規な転位反応」である本反応には一般性があること,反応機構はベックマン転位とは全く異なることから,ハロゲンと窒素原子(窒素カチオン)が一時的に結合を作る新規反応機構を提唱するに至った[Fig. 20(c)].
ペリ位の置換基をヨウ素にした場合,対応するオキシムの酸素原子をトシル酸クロライドを用いて酸素–トシル化すると,目的の酸素–トシルオキシムは生成せず,新たな物質の生成が認められた.生成物は転位生成物であるラクタムではない,各種スペクトルデータ及び元素分析データの結果,ヨウ素とオキシム窒素原子が結合形成した化合物であることが判明した(Fig. 21).この化合物をTFAの中に加えても,一部ケトン体が生成するほかには転位先生物は一切得られず,回収された.対アニオンイオンを一部交換して単結晶を得て,X線結晶構造解析により,構造を確定し,一過的な中間体として提唱してきたハロゲン–窒素結合の生成を支持することがわかった.N–I結合は原子間距離から共有結合が形成されていることがわかった.28)

このように,本研究はハロゲン原子と窒素カチオンが相互作用し一時的なハロゲン–窒素結合を形成する新しい隣接基関与の存在を世界に先駆けて明らかにした.本研究結果は基礎化学における「隣接基関与」の普遍性に確信をもたらした.
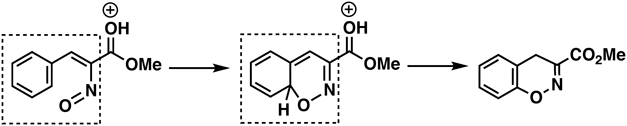
酸存在下,ニトロ基の酸素原子がベンゼン環上に環化する反応を見い出した(Fig. 22).この反応はベンゼン環が参加する1,3双極子環化付加反応を起こしていることを突き止めた.このようにベンゼン環が参加する初めての例である.

強ブレンステッド酸触媒を用いたメチル3-アリール-2-ニトロプロピオネートの分子内環化反応により4H-1,2-ベンゾオキサジンが生成することを見い出した(Fig. 22, above).29,30)この反応は,分子内のニトロ基に酸素原子が由来する芳香環の酸素官能基化反応とみなすことができる.つまりベンゼンからフェノールを作る反応に対応する.
C–H結合に酸素原子を直接導入することは非常に魅力的なアイデアだが,芳香族及び脂肪族C–H結合のいずれにおいても,その実現性はまだ非常に限られている.特に,ニトロ基の還元による芳香族窒素官能基化はよく知られているが,ニトロ基の酸素原子を酸素源として利用する研究はほとんど行われていない.
特筆すべきことは,本環化反応はベンゼン環上に電子吸引性基が存在すると反応収率がよくなり,逆に電子供与基があると反応収率は低くなる点である.これは強酸触媒で多く起きる芳香族求電子置換反応の芳香環上の置換基の傾向と全く逆であり,本反応が単なる芳香族求電子置換反応ではないことを示している.
ベンゼン存在下でのニトロオレフィンの酸触媒によるFriedel–Crafts型環化反応により4H-1,2-ベンゾオキサジンが生成することを以前報告しており,31)この反応はニトロ基からベンゼン環への分子間酸素移動反応の最初の例とみなすことができる.このように,ニトロ基からの酸素官能基導入反応は,その特異な性質から非常に興味深いが,ほとんど研究がされていなかった.
Figure 23に示すように,ベンジル位の脱プロトン化とアシニトロの水の脱離で共役ニトロソ化合物ができ,6π電子環状反応によって環化生成物ができると考えるのが一番理解しやすいが,重水素を用いた実験で,ベンジル位の脱プロトン化は起きないことがわかった.30)
結果的に,O-プロトン化アシ-ニトロ種のベンゼン環への1,3-双極子環化反応が妥当であると結論した(Fig. 22, below).三環式[3+2]環化付加体は,再芳香族化後にC–H及びC–C結合の開裂を伴って4H-1,2-ベンゾオキサジン構造を生成すると考えられる.一方,酸性度–反応速度プロファイルにおいて,過剰量の強酸であるトリフルオロメタンスルホン酸(trifluoromethanesulfonic acid: TfOH)が必要であること,エステルカルボニル基のO-プロトン化が関与していることから,反応性の高いジカチオンの寄与が考えられる(Fig. 22, below).計算による活性化エネルギーが比較的低くジカチオン環化遷移構造の振動数解析では,C–C結合形成がC–O結合形成に先行し,環化反応の一部が非同期的である可能性が示唆された.30)
5-2. 有機触媒を用いるモノカチオン性求電子体の生成5-2-1. アミノカルボン酸の芳香族アシル化反応安息香酸(1)とベンゼンをTfOH中,o-サリチル酸メチルから成るリン酸トリエステル(A)存在下で室温,20分間反応させると,芳香族ケトン(2)が収率92%で得られた(Fig. 24).32,33) Aを加えないと20分間反応しても目的の芳香族ケトン(2)は得られなかった.反応時間を24時間まで延長したところ,A非存在下でも芳香族ケトン(2)が生成したが,収率は48%に留まった.このように,リン酸トリエステル(A)は安息香酸(1)とベンゼンの反応を促進する有機触媒とみなすことができることが示唆された.さらにずっと弱い酸であるTFAを用いると,Aが存在しても目的の芳香族ケトン(2)は生成せず,エステル3が生成した.このように,芳香族アシル化反応には強酸が必要である.

また,オルト位にアミノ基を有するアントラニル酸(4)も基質として利用可能である.Aが存在しないと,TfOH中のアントラニル酸(4)は24時間反応後でも収率7%未満でしかケトン5を生成しないが,Aの存在下では20分の反応で88%の収率でケトン5が得られた(Fig. 24).TfOHに弱酸であるTFAを混合して酸度を下げると(酸度関数H0=−11.8),芳香族ケトン(5)の収率は10%に低下し,サリチル酸メチルとアントラニル酸の反応によりエステル(6)が38%の収率で形成された.さらに酸度を下げると(TFAのみ;H0=−2.7)芳香族ケトン(5)は生成せず,Aを99%の収率で回収した.
これらの反応は,Fig. 25に示すように,アントラニル酸からアシリウムカチオンを生成することが困難であるためである.芳香族アミンは酸性媒体中で十分に塩基性であり,その後のカルボン酸官能基のアシリウムカチオンへのイオン化は電荷–電荷反発により阻害される(Fig. 25).このように,リン酸トリエステルAのアシリウムカチオンへのイオン化促進効果が重要な役割を担っている.つまり,通常は求電子性を上昇させるジカチオンの生成であるが,この反応では反応の進行を抑制している.アンモニウムカチオン(NH3+)の陽電荷を中和しないと,反応中心に陽電荷を持った反応活性種(アシルカチオン)が生成しない.

様々なリン酸エステルの反応性を検討した(Fig. 26).34)有機触媒中のo-サリチル酸メチル(7)と4-ヒドロキシ安息香酸メチル(8)の組み合わせ違いは,TfOH中で安息香酸(1)とベンゼンの芳香族アシル化反応によりケトン2を与える反応の促進活性を変えた.有機触媒中のパラ異性体(8)の数がA→B→C→Dの順に増加すると,ケトン(2)の化学収率は低下し,すなわち反応促進作用が減少した(Fig. 26).トリ-p-アイソマー(D)は,芳香族アシル化の促進効果を示さなかった.

芳香族ケトン(2)の収率は,リン酸エステル触媒が1つのo-サリチル酸メチルから成る(C)ではなく,2つの(B)又は3つの(A)o-サリチル酸メチルを含む場合に大きく増加した.すなわち,強力な脱離基であるo-サリチル酸メチルのオルトエステル基が,反応基質のカルボン酸1とのP–O結合形成を促進するように機能し,エステル交換に至りアシルリン酸9が生成した(Fig. 27).

アシルリン酸自体の高い反応性を理解するため,芳香環に置換基を持たないフェノールをリン酸リンカーとしてアシルリン酸(10, 12)を合成し,強酸中のベンゼンとの反応を検討した(Fig. 28).目的の芳香族ケトン(2)を81%の収率で得た.この実験結果は,リン酸トリエステル(A)で得られた結果と同様であり,アシルリン酸エステル(10)自体が高い反応性を持ち,強酸性条件下で芳香族化合物と素早く反応して芳香族ケトンを生成することが示唆された.同様の反応条件下で,アントラニル酸部位を含むアシルリン酸塩(12)はTfOH中でベンゼンと反応したが,ケトン(5)の化学収率は中程度(収率40%)にとどまった.これは,推定中間体9(収率63%)及びリン酸トリエステルA(収率92%)の結果とは対照的である.

アントラニル酸のアシルリン酸塩の場合,生成するカチオンの電荷–電荷反発にもかかわらず,アシルリン酸塩の反応性が高い理由は,その高い開裂能にあると考えられる(Fig. 29).

カルボン酸(14),サリチル酸メチルを脱離基とするエステル(15),アシルリン酸(66)からそれぞれのアシルイオンを生成する反応プロファイルをDFT計算により検討した.アントラニル酸の場合,アミノ窒素とカルボニル酸素の両方でジプロトン化が起こる可能性がある.しかし,電荷–電荷反発のため,アシリウムカチオンを生成するための脱水反応は起こらない(Fig. 29).
出発構造の候補の1つは,分子内で水素結合した14である(Fig. 29).もう1つの候補は,サリチル酸メチルのエステル(15)であり,この場合,サリチル酸メチル内で分子内水素結合を形成することができる.この場合,生成するアシリウムカチオンの電荷–電荷反発を避けるため,プロトンの近傍にカウンターアニオンが配置された.14と15の反応プロファイルをアシルリン酸16の反応プロファイルと比較した(Fig. 29).アシルリン酸16の場合,活性化エネルギーは低く,特にTS–C構造では水素結合ネットワークによって安定化されていると思われる.アシルリン酸エステルの高い反応性,ひいてはリン酸ジエステルの高い開裂能力は,リン酸ジエステル内の共鳴効果という観点から説明することができる.19においてプロトン化(N–H+)(共有結合)が水素結合(N-----H+)になることによって電荷–電荷反発(Fig. 25)が軽減され,アシルカチオンが生成した.34)
筆者らは,窒素原子を含む,いくつかの化学結合の活性化を見い出した.これらの活性化された化学結合は,機能ある物質,特に生物活性のある分子の創製につながった.新しい機能を発現するには新しい構造が必要であり,逆に新しい構造は新しい機能を創り出すことが可能である.このような化学研究及び機能研究の連携は薬学研究において今後益々重要さを増すにちがいない.
本総説で述べた成果は名古屋市立大学薬学部並びに東京大学大学院薬学系研究科の多くの大学院生,学部生,また共同研究先の先生方・大学院生の皆様との研究の結果でありここに深く感謝申し上げます.特に多くの研究の初期から一緒に研究を行って頂きました尾谷優子博士(東京大学大学院薬学系研究科准教授)に深謝いたします.研究を自分の頭で考える面白さを教え頂いた首藤紘一先生(東京大学薬学部名誉教授),太田敏晴先生(元東京大学薬学部助教授・第一三共)に心より感謝申し上げます.また計算化学を始めるに当たって計算機センターの使い方や計算についてご指導頂きました板井明子先生(元東京大学薬学部客員教授・医薬分子設計研究所)に感謝申し上げます.残念なことに,三先生とも鬼籍に入られてしまいましたがご恩を忘れることはありません.最後になりましたが,学会賞に推薦して頂きました夏苅英昭先生[新潟薬科大学客員教授(元帝京大学薬学部教授・元東京大学薬学部教授)]に感謝申し上げます.
開示すべき利益相反はない.
本総説は2022年度日本薬学会賞の受賞を記念して記述したものである.