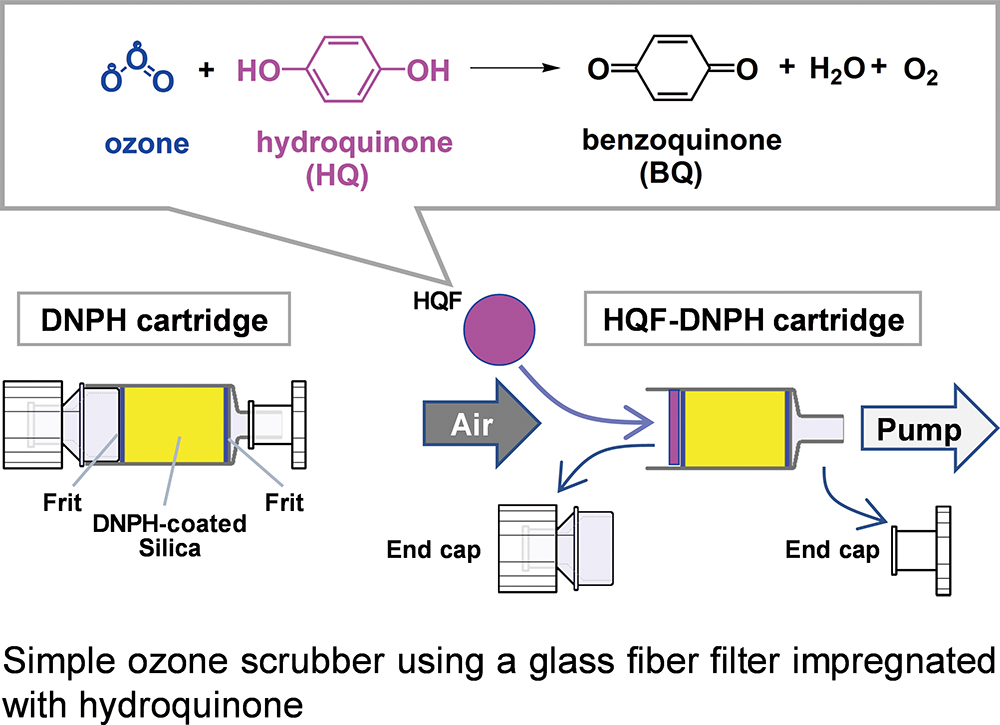
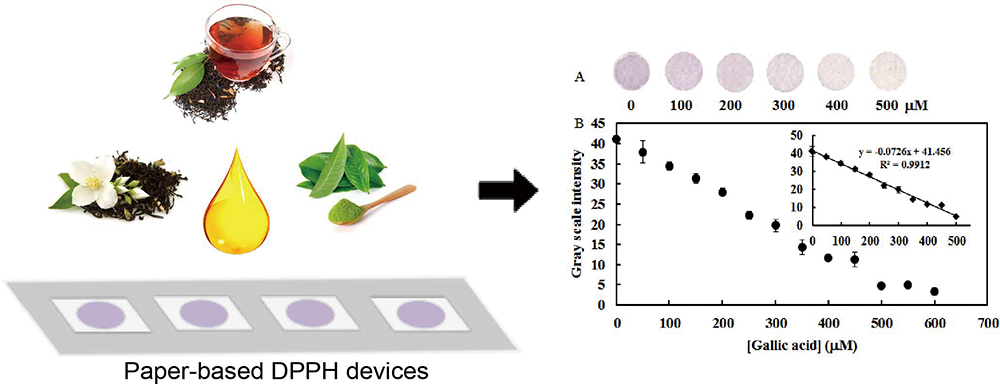
Analytical Sciences
Notice to authors (Sep. 29, 2021):
Our society has made a contract with Springer Nature to publish Vol. 38 (2022) of Anal. Sci. or later on a website of Springer Nature. The editorial board will be kept on the Japan Society for Analytical Chemistry even after the change of the publisher. If you submit a manuscript to Anal. Sci. on January 1, 2022, or later, please visit a brand new website, which will be introduced here as soon as possible. The collected manuscripts will be reviewed through the same editorial board of Anal. Sci. as before.
Until the end of December 31, 2021, this website works for receiving your manuscript as always. Please note that, however, the accepted manuscripts will be disclosed on Springer Nature's new website for both "Advance Publication" and "Publication in the final format."
Of another note is that the publication will change from "Free Access" to "Open Access." Thus far, the accepted papers have long been disclosed free of charge. From now on, the accepted papers will be disclosed on the "Open Access" basis, which is strictly opened to private and institutional subscribers of Springer Nature. If you pay the article processing charge (APC) to the publisher, the paper will fully be disclosed to all the viewers.
Additional details will be announced as soon as we have.
The Editor
Takashi Hasegawa
******
Analytical Sciences is an international journal published monthly since 1985 by the Japan Society for Analytical Chemistry (JSAC). The journal publishes papers from all areas of the analytical sciences; including fundamental and applied analytical chemistry, chemical, physical and biological analyses, and nano- and micro-analytical methodology.
・2020 Impact Factor: 2.081 [2020 Journal Citation Reports (Clarivate Analytics, 2021)]
Read more
Our society has made a contract with Springer Nature to publish Vol. 38 (2022) of Anal. Sci. or later on a website of Springer Nature. The editorial board will be kept on the Japan Society for Analytical Chemistry even after the change of the publisher. If you submit a manuscript to Anal. Sci. on January 1, 2022, or later, please visit a brand new website, which will be introduced here as soon as possible. The collected manuscripts will be reviewed through the same editorial board of Anal. Sci. as before.
Until the end of December 31, 2021, this website works for receiving your manuscript as always. Please note that, however, the accepted manuscripts will be disclosed on Springer Nature's new website for both "Advance Publication" and "Publication in the final format."
Of another note is that the publication will change from "Free Access" to "Open Access." Thus far, the accepted papers have long been disclosed free of charge. From now on, the accepted papers will be disclosed on the "Open Access" basis, which is strictly opened to private and institutional subscribers of Springer Nature. If you pay the article processing charge (APC) to the publisher, the paper will fully be disclosed to all the viewers.
Additional details will be announced as soon as we have.
The Editor
Takashi Hasegawa
******
Analytical Sciences is an international journal published monthly since 1985 by the Japan Society for Analytical Chemistry (JSAC). The journal publishes papers from all areas of the analytical sciences; including fundamental and applied analytical chemistry, chemical, physical and biological analyses, and nano- and micro-analytical methodology.
・2020 Impact Factor: 2.081 [2020 Journal Citation Reports (Clarivate Analytics, 2021)]
Published by
The Japan Society for Analytical Chemistry
10,013 registered articles
(updated on May 05, 2024)
(updated on May 05, 2024)
Online ISSN : 1348-2246
Print ISSN : 0910-6340
ISSN-L : 0910-6340
Print ISSN : 0910-6340
ISSN-L : 0910-6340
1.967
2021 Journal Impact Factor (JIF)
2021 Journal Impact Factor (JIF)