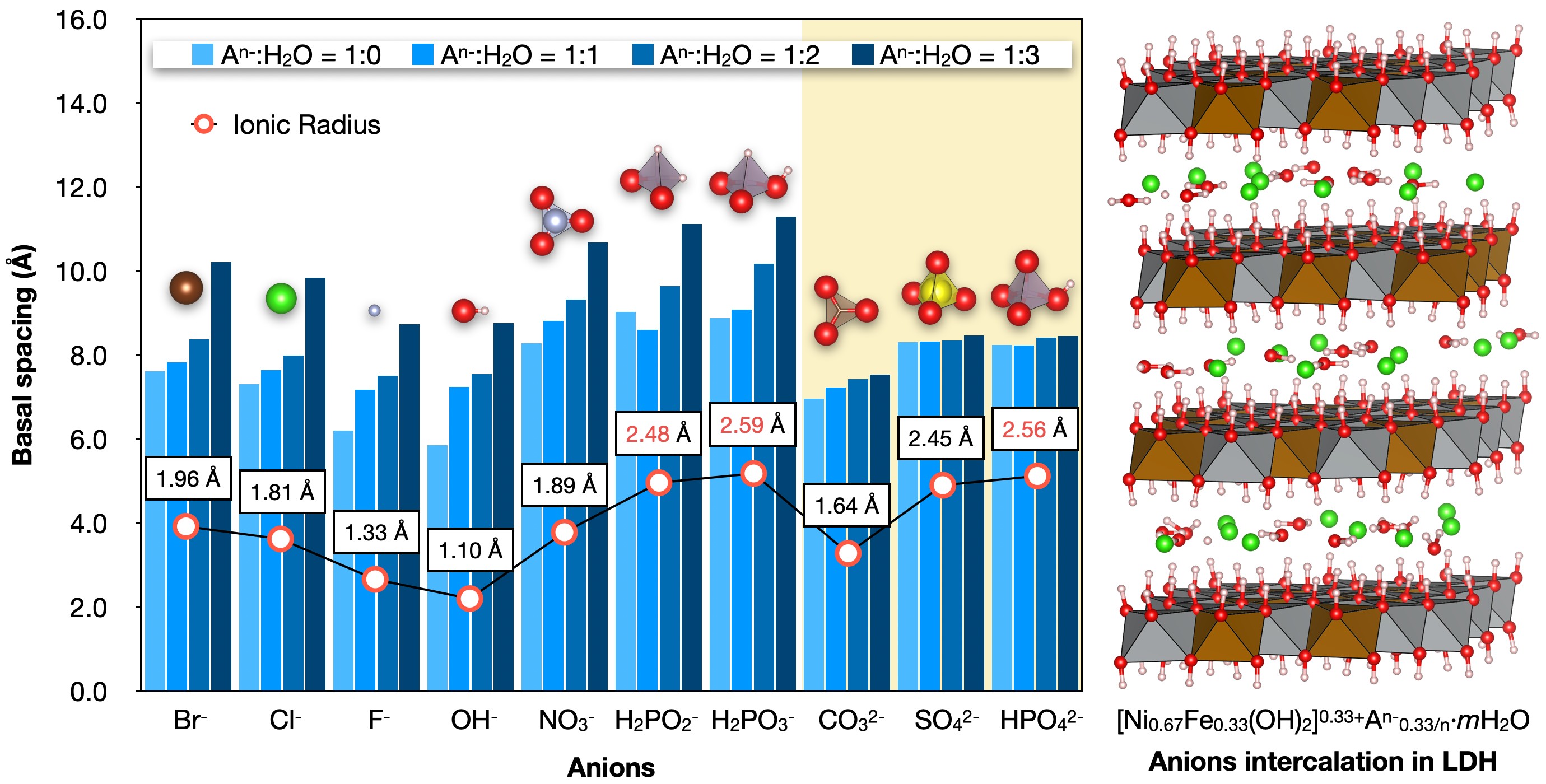
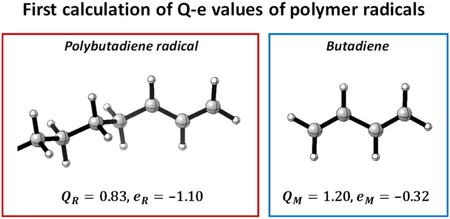
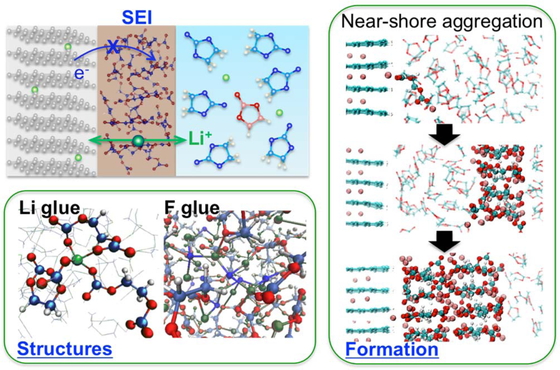
Journal of Computer Chemistry, Japan
Published by
Society of Computer Chemistry, Japan
1,137 registered articles
(updated on July 27, 2026)
(updated on July 27, 2026)
Online ISSN : 1347-3824
Print ISSN : 1347-1767
ISSN-L : 1347-1767
Print ISSN : 1347-1767
ISSN-L : 1347-1767