Abstract
G-protein coupled receptor 43 (GPR43) serves as a receptor for short-chain fatty acids (SCFAs), implicated in neutrophil migration and inflammatory cytokine production. However, the intracellular signaling pathway mediating GPR43 signaling remains unclear. Here, we show that β-arrestin 2 mediates the internalization of GPR43 by agonist. Agonism of GPR43 reduced the phosphorylation and nuclear translocation of nuclear factor-κB (NF-κB), which was relieved by short interfering RNA (siRNA) of β-arrestin 2. Subsequently, mRNA expression of proinflammatory cytokines, interleukin (IL)-6 and IL-1β, was downregulated by activation of GPR43 and knockdown of β-arrestin 2 recovered the expression of the cytokines. Taken together, these results suggest that GPR43 may be a plausible target for a variety of inflammatory diseases.
The short chain fatty acids (SCFAs) such as acetate (C2), propionate (C3) and butyrate (C4) are the major metabolic products of anaerobic bacteria fermentation in the colon.1,2) SCFAs have been reported to decrease immune-related gene expression and cytokine release by inhibiting nuclear factor-κB (NF-κB) activity,3,4) implicating SCFAs to be important molecules for treating inflammatory disorders. G-protein coupled receptor 43 (GPR43, also known as free fatty acid receptor 2, FFAR2) is one of the receptors for SCFAs and highly expressed in intestine, adipocytes and neutrophils implying its potential involvement in metabolism and immune regulation.5–7) Recently, the reports using GPR43 knockout mice in an inflammatory bowel disease (IBD) model (dextran sulfate sodium-induced colitis mouse model) have suggested that GPR43 may be a novel therapeutic target for IBD, arthritis, asthma, and other inflammatory diseases.8,9) It should be noted, however, that loss-of-function mutation of GPR43 can either block9) or enhance8) neutrophil infiltration into inflamed tissue and intestinal damage. The molecular mechanisms of the inflammatory events via modulating GPR43 remain unclear.
Generally, the ligand-activated G-protein coupled receptor (GPCRs) are phosphorylated and bound to β-arrestins (β-arrestin 1 and β-arrestin 2), leading to the internalization and desensitization of GPCRs.10) Interestingly, recent studies found that β-arrestins may play an important role in regulating NF-κB pathway and inflammation.11–13) However, the relationship between GPR43 and β-arrestins has not yet been fully characterized.
Here, we demonstrate that agonist-induced GPR43 interact with β-arrestins, with preference for β-arrestin 2 (βarr2). In addition, knockdown of βarr2 reduces the internalization of GPR43 after agonist ligation. Furthermore, the inhibition of NF-κB and downregulation of its downstream genes, interleukin (IL)-1β and IL-6, by GPR43 agonist are significantly compromised by the knockdown of βarr2. These results collectively suggest GPR43 is a promising target for inflammatory diseases via the regulation of NF-κB activity.
MATERIALS AND METHODS
Cell Preparation and CultureHeLa (CCL-2) and HEK293 (CRL-1573™) were purchased from the American Type Culture Collection (U.S.A.). Cells were grown in Dulbecco’s modified Eagle’s medium (HyClone, U.S.A.) supplemented with 10% fetal bovine serum (Gibco, U.S.A.) and 100 units/mL penicillin plus 100 µg/mL streptomycin at 37°C, in a humidified 5% CO2 atmosphere.
Synthesis of Phenylacetyl Aminothiazole (PAAT)PAAT ((S)-2-(4-chlorophenyl)-N-(5-fluorothiazol-2-yl)-3-methylbutanamide) was synthesized according to its original synthesis article.14,15)
Construction of Mammalian Expression VectorsHuman GPR43 was amplified from human cDNA library and cloned into pcDNA3.1-CLuc containing C-terminal fragment of firefly luciferase (aa. 1–416). Rat β-arrestin 1 (βarr1) and β-arrestin 2 (βarr2) cDNAs were purchased from Addgene U.S.A. (Addgene plasmid 1469316)) and cloned into pcDNA3.1-NLuc containing N-terminal fragment of firefly luciferase (aa. 398–550). Polymerase chain reaction (PCR) primers were synthesized by Bioneer, Korea. All clones were verified by sequencing (Solgent, Korea).
TransfectionTo measure the complemented luciferase activity, HEK293 cells were plated 1×104 cells in 96-well plates and the plasmids were transfected by Lipofectamine2000 (Invitrogen, CA, U.S.A.) according to the manufacturer’s instruction. Short interfering RNAs (siRNAs) against human βarr1 and βarr2 (ON-TARGET plus SMARTpool) were purchased from Dharmacon (ThermoFisher Scientific, U.S.A.). For siRNA transfection, HeLa cells were plated in 6-well plates (60–70% confluence) and then transfected with Lipofectamine RNAiMAX (Invitrogen, U.S.A.) according to the manufacturer’s instruction.
Luciferase Reporter AssayLuciferase activity was determined by OneGlo Luciferase Assay kit (Promega, U.S.A.). Dimethyl sulfoxide (DMSO) was used as a vehicle control. Significance was determined by Student’s t-test, and differences were considered significant when p<0.01.
Evaluation of mRNA Expression LevelsTotal RNA was isolated with TRIzol reagent (Invitrogen, U.S.A.), and first strand cDNA was synthesized with Omniscript Reverse Transcriptase (Qiagen, CA, U.S.A.). SYBR green-based quantitative PCR amplification was then performed with CFX 96 Real-time reverse transcription (RT)-PCR Detection system (Bio-Rad, U.S.A.) and the SYBR Green Master Mix (Bio-Rad, U.S.A.). All reactions were run in triplicate, and data were analyzed by the 2−ΔΔCT method. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) amplification was used as the control. Significance was determined with GAPDH-normalized 2−ΔΔCT values.
Western Blot AnalysisThe cells were transfected with siRNA for 24h, and then treated with 5 µM PAAT for 30 min. Proteins were homogenized in ice-cold buffer consisting of 50 mM Tris–HCl (pH 8.0), 5 mM ethylene glycol bis(2-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EDTA), 150 mM NaCl, 1% NP-40, 0.1% sodium dodecyl sulfate (SDS), 1 mM phenylmethylsulfonyl fluoride (PMSF), and one protease inhibitor cocktail tablet (Roche, Germany). The nuclear proteins were extracted with the NucBuster Protein Extraction kit (Novagen, Germany). Anti-HA, anti-Myc, anti-GAPDH and anti-histone deacetylase (HDAC1) antibodies were purchased from Santa Cruz Biotechnology, U.S.A. Anti-βarr2, anti-phospho-Ser536-NF-κB and anti-NF-κB antibodies were obtained from Cell Signaling Technology, U.S.A. Anti-βarr1 antibody was purchased from Epitomics, U.S.A. The protein bands were visualized by LAS-4000 luminescent image analyzer and Multi Gauge software, version 3.0 (FUJIFILM, Japan).
ImmunoprecipitationFor co-immunoprecipitation (co-IP), the cells were extracted in ice-cold Nonidet P40 extraction buffer (50 mM Tris–HCl, pH 7.5, containing 1 mM EDTA, 120 mM NaCl, 1% NP-40). An equal amount of each protein lysate was incubated with anti-Myc monoclonal antibodies for 3 h at 4°C, followed by incubation with 30 µL of protein G-Agarose beads (Santa Cruz Biotechnology, U.S.A.) for 3 h. The immune complexes were washed three times with lysis buffer and then boiled in Laemmli SDS sample buffer for 5 min. Immunoreactive signals were detected with enhanced chemiluminescence (ECL) kit and visualized by LAS-4000.
Establishment of Stable Cell Line for High-Content AssayHuman GPR43 fused to GFP was cloned into pIRESpuro3 (Clontech, U.S.A.) vector and transfected into HeLa cells by Lipofectamine 2000 (Invitrogen, U.S.A.) according to the manufacturer’s protocols. The transfected cells were selected by using 1 µg/mL of puromycin for 2 weeks and the colonies were individually cloned and proliferated.
Cells were plated in a 96-well plate at 5×103 cells/well. After 24 h, nuclei were stained with Hoechst 33342(1 µg/mL; Invitrogen, U.S.A.) for 5 min and then spot formation of GPR43-GFP signals were analyzed using Spot Detector BioApplication assay that was available in the Cellomics® ArrayScan® VTI HCS reader (ThermoFisher, U.S.A.).
Statistical AnalysisStatistical significance was determined by analysis of variance by the Student’s t-test. Results were considered significant at p values less than 0.05 and are labeled with a single asterisk. In addition, p values less than 0.01 and less than 0.001 are designated with double and triple asterisks, respectively.
RESULTS
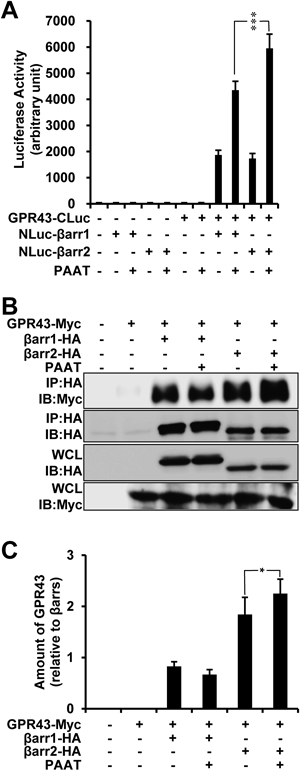
GPR43 Interacts with β-ArrestinsGPR43 has been implicated in neutrophil migration and inflammatory process, however, little is known about underlying molecular mechanisms. Other than heterotrimeric G protein, GPCRs can be coupled to and negatively regulated by β-arrestins.17) The role of β-arrestins in GPR43 signaling, however, has not yet been investigated. To elucidate the interaction of GPR43 with β-arrestins, we generated the systems measuring protein-protein interaction (PPI) between GPR43 and β-arrestins using bimolecular luminescence complementation method (BiLC) (see in Materials and Methods). The GPR43-fused to C-terminal region of firefly luciferase (GPR43-CLuc) and/or βarr1 or βarr2 fused to N-terminal region of firefly luciferase (NLuc-βarr1/2) were transiently transfected in HEK293 cells. Co-expression of GPR43 and βarr1 or 2 resulted in the complemented luciferase activity, which was augmented by PAAT treatment (Fig. 1A). Notably, βarr2 coupled to GPR43 showed more activity compared to βarr1 (Fig. 1A, lanes 9 and 11). To further determine the association between βarr1/2 and GPR43, we performed co-immunoprecipitation by transfecting plasmids encoding myc tagged-GPR43, HA tagged-βarr1, and HA tagged-βarr2 in HEK293 cells. Figures 1B and C showed that GPR43 was co-precipitated with β-arrestins. It is of note that βarr2 more strongly interacts with GPR43 in terms of its expression level. Moreover, the amount of GPR43 coupled to βarr2 was increased by PAAT (Fig. 1B, lane 6, and Fig. 1C, lane 6). Taken together, we demonstrated that GPR43 associates with β-arrestins, especially βarr2.
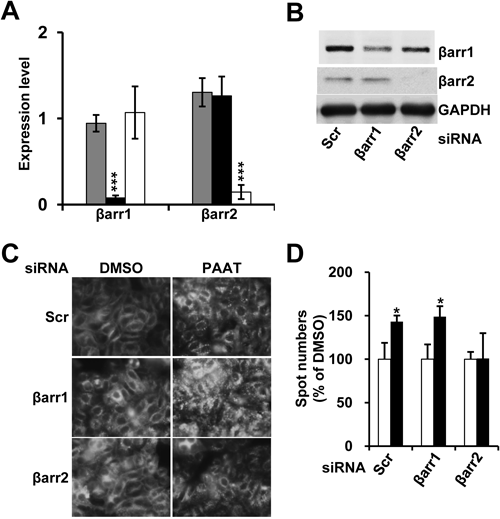
It has been well known that β-arrestins induce the internalization of GPCRs via clathrin-coated pits and this is the one of the important mechanisms to desensitize GPCR signaling.10) In order to investigate the relationship between β-arrestins and GPR43, we generated the stable cell line expressing GPR43 tagged with GFP in HeLa cells. First, we confirmed the efficiency of siRNAs of βarr1 and βarr2 in terms of mRNA level (Fig. 2A) and protein level (Fig. 2B) in HeLa cell line expressing GPR43-GFP. To visualize and quantitatively analyze the internalization of GPR43, we used ArrayScan™ (Cellomics™, Pittsburgh, PA), a microtiter plate imaging system that permits cellular quantitation of fluorescence in whole cells. After the transfection of siRNAs, the cells were treated with PAAT or vehicle, and then fixed. The cell images were acquired and analyzed quantitatively using Cellomics BioApplication analysis software (Spot Detector) through spot formation of internalized GPR43-GFP. As shown in Fig. 2C, the number of intracellular GFP spots in the scrambled or βarr1 siRNA-transfected cells was significantly increased by the addition of PAAT (Fig. 2C and quantified in Fig. 2D). However, in the PAAT-induced cells transfected with βarr2 siRNA, the spot number of internalized GPR43 was almost not altered compared to control (Figs. 2C, D). These results are consistent with above results that βarr2 interacts to GPR43 with higher affinity that βarr1 (Fig. 1), suggesting the agonist-induced internalization of GPR43 is mainly mediated by βarr2.
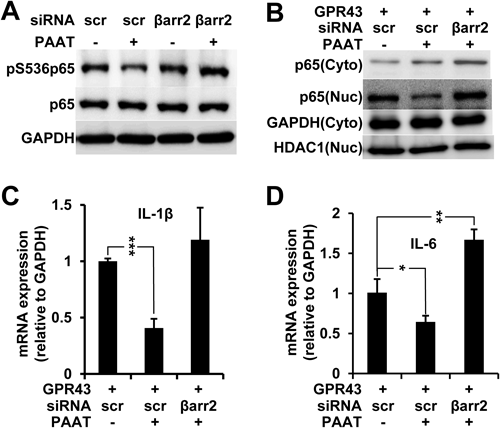
Previous studies have shown that GPR43 is necessary for the migration of neutrophil and to maintain normal condition during inflammation response in the animal models of colitis, arthritis and asthma.8,9) To elucidate the effect of GPR43 in inflammatory process, we determined whether GPR43-βarr2 signaling can modulate the activity of NF-κB. The cells stably expressing GPR43-GFP were transfected with scrambled siRNA or βarr2 siRNA. Forty-eight hours after transfection, the cells were treated with or without PAAT, and then lysed. The phosphorylation of p65 subunit of NF-κB at Ser 536 is one of the good indicators that show the activation status of NF-κB.18) It is of note that the activation of GPR43 by the addition of PAAT compromised the phosphorylation of p65 (Fig. 3A, lane 2). Surprisingly, the knockdown of βarr2 restored the phosphorylation at Ser536 of p65 in the presence of PAAT (Fig. 3A, lane 4), implicating that βarr2 mediates GPR43 signaling to NF-κB. To further confirm this result, we fractionated the cells transfected with GPR43 and/or βarr2 siRNA into the cytoplasm and nucleus. PAAT blocked the nuclear translocation of NF-κB and knockdown of βarr2 restored the level of nuclear NF-κB (Fig. 3B), which is consistent with the result in Fig. 3A. To investigate the cellular consequences of GPR43-βarr2-NF-κB axis, we performed quantitative real time RT-PCR for the quantitation of the expression of inflammatory cytokines such as IL-1β and IL-6, both of which are the targets of NF-κB. PAAT-treated cells showed a marked decrease in mRNA levels of those cytokines, however, expression of IL-1β (Fig. 3C) and IL-6 (Fig. 3D) were restored by the knockdown of βarr2, clearly indicating that GPR43 negatively regulates inflammatory cytokines by the modulation of NF-κB activity through βarr2.
DISCUSSION
GPR43 has been implicated in inflammatory processes such as neutrophil migration and cytokine production,8,9,19–22) but its molecular mechanisms have not been characterized. Here, we provided direct evidences for the first time that βarr2 specifically interact with GPR43 and mediate the signaling to NF-κB and subsequently cytokine production. In general, an agonist-activated GPCR is phosphorylated by G protein-coupled receptor kinases (GRKs) and subsequently bound to β-arrestins, which regulate desensitization, internalization, intracellular signaling, and recycling of GPCRs.10) Some GPCRs interact with equal affinity with βarr1 and βarr2, others prefer either of them, usually βarr2.23) Based upon this general phenomenon, the assay methods measuring the activity of GPR43 have been developed such as PathHunterTM 24) and Bioluminescence Resonance Energy Transfer (BRET)25) technology, both of which utilized βarr2. They did not, however, provide direct evidence that GPR43 interacts with βarr2. Our results clearly demonstrated that βarr2 is more strongly associated with GPR43 in an agonist-dependent manner than βarr1 (Fig. 1) and specifically mediates the internalization of GPR43 (Fig. 2).
In addition to the roles of desensitization and internalization of GPCRs, several studies unveiled the novel function of β-arrestins in NF-κB signaling. It has been shown that βarr2 directly binds to and blocks phosphorylation and degradation of Inhibitor of NF-κB (IκB), leading to the inhibition of the activity of NF-κB.12,26) In addition, upstream kinases of IκB such as IκB kinases (IKKs) and NF-κB-inducing kinase (NIK) interact with β-arrestins.13) Moreover, TRAF6, further upstream component of NF-κB signaling, also associates itself with β-arrestins in a stimulus-dependent manner.27) Recently, it has been shown that GPR120, a receptor for long-chain fatty acids and omega-3 fatty acid, sequesters transforming growth factor β-activated kinase 1 (TAK1) binding protein 1 (TAB1) from TAK1 complex, subsequently leading to the inhibition of NF-κB and inflammation.28) Collectively, it is hard to discern the exact mechanism that enables βarr2 to regulate NF-κB signaling. GPR43-βarr2 signaling axis may utilize one of the above mechanisms and needs to be further investigated. In addition, it will be helpful to unveil the relationship between GPR40, GPR41, and β-arrestins to compare the signaling cascade between the free fatty acid receptor family.
In conclusion, we demonstrated that GPR43 modulates NF-κB activity via βarr2. As the agonists of GPR43 are known to elicit neutrophil migration through pertussis toxin-sensitive p38 mitogen activated protein (MAP) kinase pathway, βarr2-biased agonists may have a potential anti-inflammatory effect without the side effect.
Acknowledgements
This research was supported by Grants from by the International Science and Business Belt Program through the Ministry of Science, ICT, and Future Planning (former Education, Science and Technology) (2012K001574) and KRIBB Research Initiative Program.
REFERENCES
- 1) Cummings JH. Short chain fatty acids in the human colon. Gut, 22, 763–779 (1981).
- 2) Wong JM, de Souza R, Kendall CW, Emam A, Jenkins DJ. Colonic health: fermentation and short chain fatty acids. J. Clin. Gastroenterol., 40, 235–243 (2006).
- 3) Segain JP, Raingeard de la Bletiere D, Bourreille A, Leray V, Gervois N, Rosales C, Ferrier L, Bonnet C, Blottiere HM, Galmiche JP. Butyrate inhibits inflammatory responses through NF-κB inhibition: implications for Crohn’s disease. Gut, 47, 397–403 (2000).
- 4) Tedelind S, Westberg F, Kjerrulf M, Vidal A. Anti-inflammatory properties of the short-chain fatty acids acetate and propionate: a study with relevance to inflammatory bowel disease. World J. Gastroenterol., 13, 2826–2832 (2007).
- 5) Senga T, Iwamoto S, Yoshida T, Yokota T, Adachi K, Azuma E, Hamaguchi M, Iwamoto T. LSSIG is a novel murine leukocyte-specific GPCR that is induced by the activation of STAT3. Blood, 101, 1185–1187 (2003).
- 6) Nilsson NE, Kotarsky K, Owman C, Olde B. Identification of a free fatty acid receptor, FFA2R, expressed on leukocytes and activated by short-chain fatty acids. Biochem. Biophys. Res. Commun., 303, 1047–1052 (2003).
- 7) Brown AJ, Goldsworthy SM, Barnes AA, Eilert MM, Tcheang L, Daniels D, Muir AI, Wigglesworth MJ, Kinghorn I, Fraser NJ, Pike NB, Strum JC, Steplewski KM, Murdock PR, Holder JC, Marshall FH, Szekeres PG, Wilson S, Ignar DM, Foord SM, Wise A, Dowell SJ. The Orphan G protein-coupled receptors GPR41 and GPR43 are activated by propionate and other short chain carboxylic acids. J. Biol. Chem., 278, 11312–11319 (2003).
- 8) Maslowski KM, Vieira AT, Ng A, Kranich J, Sierro F, Di Yu, Schilter HC, Rolph MS, Mackay F, Artis D, Xavier RJ, Teixeira MM, Mackay CR. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature, 461, 1282–1286 (2009).
- 9) Sina C, Gavrilova O, Forster M, Till A, Derer S, Hildebrand F, Raabe B, Chalaris A, Scheller J, Rehmann A, Franke A, Ott S, Hasler R, Nikolaus S, Folsch UR, Rose-John S, Jiang HP, Li J, Schreiber S, Rosenstiel P. G protein-coupled receptor 43 is essential for neutrophil recruitment during intestinal inflammation. J. Immunol., 183, 7514–7522 (2009).
- 10) Luttrell LM, Lefkowitz RJ. The role of beta-arrestins in the termination and transduction of G-protein-coupled receptor signals. J. Cell Sci., 115, 455–465 (2002).
- 11) Shenoy SK, Lefkowitz RJ. Multifaceted roles of beta-arrestins in the regulation of seven-membrane-spanning receptor trafficking and signalling. Biochem. J., 375, 503–515 (2003).
- 12) Gao H, Sun Y, Wu Y, Luan B, Wang Y, Qu B, Pei G. Identification of beta-arrestin2 as a G protein-coupled receptor-stimulated regulator of NF-kappaB pathways. Mol. Cell, 14, 303–317 (2004).
- 13) Witherow DS, Garrison TR, Miller WE, Lefkowitz RJ. β-Arrestin inhibits NF-κB activity by means of its interaction with the NF-κB inhibitor IκBα. Proc. Natl. Acad. Sci. U.S.A., 101, 8603–8607 (2004).
- 14) Lee T, Schwandner R, Swaminath G, Weiszmann J, Cardozo M, Greenberg J, Jaeckel P, Ge H, Wang Y, Jiao X, Liu J, Kayser F, Tian H, Li Y. Identification and functional characterization of allosteric agonists for the G protein-coupled receptor FFA2. Mol. Pharmacol., 74, 1599–1609 (2008).
- 15) Wang Y, Jiao X, Kayser F, Liu J, Wang Z, Wanska M, Greenberg J, Weiszmann J, Ge H, Tian H, Wong S, Schwandner R, Lee T, Li Y. The first synthetic agonists of FFA2: Discovery and SAR of phenylacetamides as allosteric modulators. Bioorg. Med. Chem. Lett., 20, 493–498 (2010).
- 16) Luttrell LM, Ferguson SS, Daaka Y, Miller WE, Maudsley S, Della Rocca GJ, Lin F, Kawakatsu H, Owada K, Luttrell DK, Caron MG, Lefkowitz RJ. β-Arrestin-dependent formation of β2 adrenergic receptor-Src protein kinase complexes. Science, 283, 655–661 (1999).
- 17) Reiter E, Lefkowitz RJ. GRKs and beta-arrestins: roles in receptor silencing, trafficking and signaling. Trends Endocrinol. Metab., 17, 159–165 (2006).
- 18) Hayden MS, Ghosh S. Signaling to NF-κB. Genes Dev., 18, 2195–2224 (2004).
- 19) Cox MA, Jackson J, Stanton M, Rojas-Triana A, Bober L, Laverty M, Yang X, Zhu F, Liu J, Wang S, Monsma F, Vassileva G, Maguire M, Gustafson E, Bayne M, Chou CC, Lundell D, Jenh CH. Short-chain fatty acids act as antiinflammatory mediators by regulating prostaglandin E(2) and cytokines. World J. Gastroenterol., 15, 5549–5557 (2009).
- 20) Vinolo MAR, Ferguson GJ, Kulkarni S, Damoulakis G, Anderson K, Bohlooly-Y M, Stephens L, Hawkins PT, Curi R. SCFAs induce mouse neutrophil chemotaxis through the GPR43 receptor. PLoS ONE, 6, e21205 (2011).
- 21) Kim S, Kim YM, Kwak YS. A novel therapeutic target, GPR43; where it stands in drug discovery. Arch. Pharm. Res., 35, 1505–1509 (2012).
- 22) Macia L, Thorburn AN, Binge LC, Marino E, Rogers KE, Maslowski KM, Vieira AT, Kranich J, Mackay CR. Microbial influences on epithelial integrity and immune function as a basis for inflammatory diseases. Immunol. Rev., 245, 164–176 (2012).
- 23) Oakley RH, Laporte SA, Holt JA, Caron MG, Barak LS. Differential affinities of visual arrestin, beta arrestin 1, and beta arrestin 2 for G protein-coupled receptors delineate two major classes of receptors. J. Biol. Chem., 275, 17201–17210 (2000).
- 24) Yin H, Chu A, Li W, Wang B, Shelton F, Otero F, Nguyen DG, Caldwell JS, Chen YA. Lipid G protein-coupled receptor ligand identification using β-arrestin PathHunter assay. J. Biol. Chem., 284, 12328–12338 (2009).
- 25) Hudson BD, Christiansen E, Tikhonova IG, Grundmann M, Kostenis E, Adams DR, Ulven T, Milligan G. Chemically engineering ligand selectivity at the free fatty acid receptor 2 based on pharmacological variation between species orthologs. FASEB J., 26, 4951–4965 (2012).
- 26) Luan B, Zhang Z, Wu Y, Kang J, Pei G. Beta-arrestin2 functions as a phosphorylation-regulated suppressor of UV-induced NF-kappaB activation. EMBO J., 24, 4237–4246 (2005).
- 27) Wang Y, Tang Y, Teng L, Wu Y, Zhao X, Pei G. Association of beta-arrestin and TRAF6 negatively regulates Toll-like receptor-interleukin 1 receptor signaling. Nat. Immunol., 7, 139–147 (2006).
- 28) Oh DY, Talukdar S, Bae EJ, Imamura T, Morinaga H, Fan W, Li P, Lu WJ, Watkins SM, Olefsky JM. GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell, 142, 687–698 (2010).
