Abstract
Alpha-lipoic acid (LA), a metabolic antioxidant, is a natural compound and its biological function has been well studied in various human diseases. The present study was designed to investigate the cytoprotective effect and the molecular mechanisms of LA in paraquat (PQ)-induced oxidative stress injury using BEAS-2B human bronchial epithelial cells. LA co-treatment prevented PQ-induced BEAS-2B cell death. LA also prevented PQ-induced increases in total reactive oxygen species (ROS), lactate dehydrogenase (LDH) and malondialdehyde (MDA). LA also increased the expression of detoxifying phase II enzyme encoding genes and antioxidant genes including HO-1, NQO1, CAT, GPX3 and GPX4, resulting in the attenuation of the decreases of antioxidants during PQ-induced oxidative stress. Nuclear factor erythroid related factor 2 (Nrf2) was induced by LA. Additionally, translocation of Nrf2 from the cytoplasm to the nucleus was promoted by LA treatment. While LA was responsible for the upregulation of Nrf2, it also activated and up-regulated the downstream proteins heme oxygenase-1 (HO-1) and reduced nicotinamide adenine dinucleotide phosphate (NAD(P)H) quinone oxidoreductase 1 (NQO1). The data collectively suggest that the beneficial effect of LA involving the activation of cytoprotective antioxidant genes make LA a potential candidate in the prevention of PQ-induced oxidative stress-related bronchial cell death, pending clinically relevant studies.
Paraquat (1,1-dimethyl-4,4′-bipyridinium chloride; PQ), is one of the most widely used herbicides worldwide and a potent reactive oxygen species (ROS) inducer. The main target organ of PQ is the lung, due to its preferential accumulation by different epithelium cells.1,2) Human bronchial BEAS-2B cells are one of the major respiratory cells in humans and are the cells exposed to incoming air in the lung. Therefore, BEAS-2B cells were judged to be a reasonable model system to explore the protective effect of antioxidant mechanisms as well as the pathogenesis of oxidative stress-induced respiratory diseases, because these cells are established from healthy human bronchi.3–5) Furthermore, BEAS-2B cells have been productively used to study the mechanisms underlying oxidative stress induced cell death.6)
ROS refer to a collection of oxygen-derived species that include oxygen radicals, such as superoxide anion radical (O2•−) and hydroxyl radicals (•OH), as well as non-radical derivatives of O2 including hydrogen peroxide (H2O2), and singlet oxygen (1O2). Under physiological conditions, ROS formation is in balance with the antioxidant capacity of the host. However, excess amount of ROS in cells can cause cellular damage and eventual cell death. Such oxidative stresses have been implicated in the pathogenesis of cell death and tissue damage.1,7,8)
Nuclear factor erythroid related factor 2 (Nrf2), nuclear factor (erythroid-derived 2)-like 2 (NFE2L2) is a prime transcription factor that governs the antioxidant defense system associated as a modifier of different lung diseases that involve oxidative stress and inflammation.9) In normal cells, the expression of Nrf2 is suppressed by kelch-like ECH-associated protein 1 (Keap1) to ensure control of ubiquitination and proteasomal degradation processing. The expression of Nrf2 can be induced by multiple stresses, antioxidants (e.g., sulforaphane) and other physiological stimuli, which will be allowed with Keap-1-mediated dissociation of Nrf2 from the repressor complex. Subsequently, the freed Nrf2 can stabilize and eventually translocate to the nucleus for gene regulation.10,11) Nrf2 thus plays a role as a transcription factor that can promptly regulate antioxidant responsive genes such as catalase (CAT), glutathione peroxidase (GPX) and superoxide dismutase (SOD1). In addition, Nrf2 also governs phase II drug metabolism genes such as heme oxygenase-1 (HO-1) and the cytosolic flavoprotein reduced nicotinamide adenine dinucleotide phosphate (NAD(P)H) quinone oxidoreductase 1 (NQO1).9,12) HO-1 is a potent detoxifying/antioxidant enzyme, which catabolizes heme to release equimolar amounts of free iron (Fe+), carbon monoxide and biliverdin. The latter is converted to bilirubin by the ubiquitous enzyme biliverdin reductase.13) Accumulating evidence is consistent with the idea that HO-1 has a pulmonary protective role. HO-1 induction in lung epithelium cells is considered an important protective mechanism against oxidative stress.14,15) NQO1 protects cells against the toxicities of quinones and their metabolic precursors by catalyzing an obligatory two-electron reduction of these compounds, as well as acting as a coenzyme Q (ubiquinone) reductase. NQO1 can facilitate the conversion of α-tocopherolquinone to α-tocopherolhydroquinone, contributing to the maintenance of this important endogenous antioxidants.14,16)
Alpha lipoic acid (LA), also designated as 1,2-dithiolane-3-pentanoic acid, is a natural dithiol compound with excellent antioxidant properties and high efficacy of chemoprotection.17) It is consumed in the daily diet of humans. Following consumption, it is absorbed through the blood–brain barrier, taken up and transformed in cells and tissues into dihydrolipoic acid (DHLA).18) It has been recognized as a potential cytoprotective candidate for the prevention of different pathologies that may be related to an imbalance of the oxidoreductive cellular status.19,20) LA has multiple pharmacological effects that are able to ameliorate lung injury.21,22) All these features support the potential of LA as a candidate compound to alleviate oxidative stress. However, the efficacy of LA in PQ-induced oxidative toxicity in human bronchial epithelial cells is unknown.
In present study, we hypothesized that LA can function as an endogenous antioxidant with chemo-protective activity to activate Nrf2 and its regulated downstream antioxidant genes. To test our hypothesis, a series of experiments were conducted and the data allow us to suggest that the induction of Nrf2 activity may contribute in protection against PQ-induced ROS stress.
Materials and Methods
MaterialsLA, PQ, 3-(4,5-dimethylthiazol-2-yl)-2, 5-diphenyl-tetrazolium bromide (MTT), Trypan blue stain solution and Triton-X100 were purchased from Sigma-Aldrich (St. Louis, MO, U.S.A.). Bronchial epithelial cell basal medium (BEBM) was obtained from Lonza (Walkersville, MD, U.S.A.). Fetal bovine serum (FBS) was purchased from HyClone (Logan, UT, U.S.A.) and antibiotic/antimycotic (100 U/mL penicillin, 100 µg/mL streptomycin and 0.25 µg/mL amphotericin B) were obtained from Gibco (Grand Island, NY, U.S.A.). Total ROS detection kit was purchased from Enzo Life Sciences (Farmingdale, NY, U.S.A.). A lactate dehydrogenase (LDH) assay kit was supplied from Roche (Pleasanton, CA, U.S.A.). Nuclear/cytosol fraction kit was provided from Biovision (Mountain View, CA, U.S.A.). Primary antibody against each Nrf2, HO-1, NQO1, Lamin B, and relevant secondary antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, U.S.A.). The primary antibody for β-actin was obtained from Abcam (Cambridge, MA, U.S.A.).
Cell Culture and Drug TreatmentThe BEAS-2B human bronchial epithelial cell line was obtained from American Type Culture Collection (Manassas, VA, U.S.A.). BEAS-2B cells were cultured in bronchial epithelial basal medium and maintained for 10 to 15 passages in 37°C humidified incubator containing 5% CO2. PQ was dissolved in distilled water as 1 m stock solution and diluted in culture medium at the indicated concentrations for different times. Ethanol was used as solvent for LA and the stock concentration was 50 mm. BEAS-2B cells were co-treated with different concentrations of PQ and LA for indicated times. The final concentration of ethanol in the media was <0.01%.
Cell Viability and MorphologyBEAS-2B cells were seeded in 96 well plates at 105 cells/well and cell viability was measured by an established MTT assay. Briefly, the cells were co-treated with indicated concentration of PQ (0–1 mm) for varying times in the continued presence of vehicle or LA (0–1 mm). After incubation, cells were exposed to 20 µL of 5 mg/mL MTT solution for 4 h at 37°C, and the medium was carefully removed by aspiration. The formazan crystals were solubilized in 100 µL of dimethylsulfoxide prior to incubation for a further 30 min. Finally, the color of solution was read at 590 nm using a VictorTM X3 multilabel reader (PerkinElmer, Waltham, MA, U.S.A.). The result was expressed as a percentage relative to the control (survival of control). In another set of experiments, following incubation of the cells with PQ and LA for 48 h, cell morphology (n=5 per group) was examined using an Axiovert 25 phase contrast light microscope (Carl Zeiss, Jena, Germany).
LDH Release AssayLDH assay is widely used to assess membrane integrity during cell death. We conducted the LDH assay using a colorimetric technique. In brief, 1×105 cells/well was seeded in 96-well plates to reach 70–80% confluency. The cells were then co-treated with target concentrations of LA and PQ for indicated concentration for varying times. At the end of treatment, 100 µL aliquots of the cell culture supernatant were transferred to wells of another 96-well plate followed by the addition of 100 µL of freshly prepared reaction mixture. Following 30 min of incubation at room temperature in the dark, the absorbance was read at 490 nm using a Victor™ X3 multilabel reader (PerkinElmer). The amount of LDH was presented as a percent compared to the total amount of LDH present in cells treated with 2% Triton-X100.
Determination of Total ROSTotal ROS were detected in BEAS-2B cells using the ROS detection kit according to the manufacturer’s protocol (Enzo Life Sciences). The assay is designed to detect total ROS production in live cells by fluorescence microscopy. The total ROS detection reagent reacts with H2O2, peroxynitrite and hydroxyl radicals. Briefly, BEAS-2B cells were cultivated in wells of 12-well tissue culture plates at a density of 1×105 cells/mL and grown to 50–70% confluency. Cells were washed twice with wash buffer and then incubated with the ROS detection solution for 1 h at 37°C in the dark. Thereafter, cells were treated with medium containing LA and PQ for 3 h. Pyocyanin and N-acetyl-l-cysteine (NAC) was used as a positive control and negative control, respectively. The medium was removed and replaced with wash buffer containing the ROS detection solution. After 20 min incubation at 37°C, cells were washed twice with wash buffer to remove any excess ROS detection solution. The fluorescence intensity was then measured using an Axiovert-25 Microscope (Carl Zeiss) with a filter range of Ex/Em: 490/525 nm. At least five regions in each culture plate were observed to determine the fluorescence intensity. The total quantity of ROS level was proportional of fluorescence intensity.
Measurement of Cellular Lipid PeroxidationAs a marker of lipid peroxidation levels, the total amounts of malondialdehyde (MDA) were measured using the lipid peroxidation quantification kit (Abcam). In principle, xenobiotics prone to cause oxidative stress, and subsequently produce lipid peroxidase, which further reacts to produce MDA. The MDA reacts with thiobarbituric acid (TBA), and generates MDA-TBA adduct that is estimated by colorimetric analysis. Briefly, cells were co-treated with 0.2 mm PQ and 0.5 mm LA in 6-well plates. After incubation, cells were lysed with 300 µL MDA lysis buffer. Two hundred microliters of supernatant was collected to quantify lipid peroxidase by centifugation for 10 min. After collecting each supernatant, 600 µL of TBA solution was dispensed in each supernatant and incubated at 95°C for 1 h. Then the mixture was cooled in an ice bath for 10 min. Finally, 200 µL of the mixture was transferred into wells of a 96-well microplate for colorimetric analysis. The absorbance was read at 530 nm using a VictorTM X3 multilabel reader (PerkinElmer). The peroxidation was counted from a standard curve using a standard MDA concentration series and expressed in terms of amount nmol/mg protein.
Total Protein Extraction and Western Blot AnalysisAfter drug treatment, cells were washed with ice cooled 1×phosphate-buffered saline (PBS) and then added to RIPA lysis buffer (Santa Cruz Biotechnology) containing a protease inhibitor cocktail (Santa Cruz Biotechnology) and dithiothreitol (DTT) to collect protein. After that, cells were collected and centrifuged at 14000×g for 15 min at 4°C. The supernatant was separated from cellular debris and the protein concentration was determined using the BCA protein assay kit (Thermo Scientific, Pittsburgh, PA, U.S.A.) with bovine serum albumin as the standard. The protein samples were mixed with 2×sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) sample loading buffer and boiled for 3 min. Fifteen micrograms of protein were separated by 4–20% gradient SDS-PAGE (Mini-PROTEAN® TGX™ Precast Gel; Bio-Rad, Hercules, CA, U.S.A.) at 100 V for 90 min. The resolved proteins were transferred onto a polyvinylidene fluoride membrane (Trans-Blot SD Semi-Dry Cell; Bio-Rad) at 15V for 1 h. Each blot was blocked with 5% skim milk-Tris buffered saline containing 0.1% Tween-20 (TBST) at room temperature for 1 h. The blots were incubated with anti-Nrf2 (1 : 500), anti-HO-1(1 : 500), anti-NQO1 (1 : 500) and anti-Lamin B (1 : 500) antibody, and anti-β-actin primary antibody was incubated in 5% fat free skim milk in PBS containing Tween-20 (PBST) overnight at 4°C. After incubation, the membranes were rinsed three times with 1×TBST and then incubated with horseradish peroxidase-conjugated secondary antibody (diluted 1 : 10000 in 5% skim milk in TBST) at room temperature for 90 min. The immunoreactive bands were detected by enhanced chemiluminescence according to the manufacturer’s instructions (Bio-Rad) and images were taken using a ChemiDoc XRS+ imaging system with Image Lab™ software (Bio-Rad).
Harvesting of Nuclear and Cytosolic ExtractsAfter treatment with LA and PQ, BEAS-2B cells (4×105 cells/mL) were rinsed twice with ice-cold 1×PBS and harvested by scraping into ice-cold 1×PBS and centrifuging at 600×g for 5 min at 4°C. Pellets were suspended in 0.2 mL CEB-A containing DTT and protease inhibitor for 10 min on ice. Then, 11 µL ice-cold cytosol extraction buffer-B was added and incubated for 1 min. The supernatant was collected by centrifugation at maximum speed for 5 min as the cytosolic extract. The remaining pellets were each suspended in 100 µL of ice-cold nuclear extraction buffer mix at 4°C for 40 min and spin down at maximum speed in a microcentifuge for 10 min. The supernatant represented the nuclear extract.
RNA Extraction and Quantitative Real-Time Polymerase Chain Reaction (PCR)After treatment of BEAS-2B cells with LA and/or PQ, total RNA was isolated using a mini RNA isolation kit (Qiagen, Valencia, CA, U.S.A.) according to the manufacturer’s instructions. Concentration and quality of total RNA were measured using a ND-1000 spectrophotometer (NanoDrop, Wilmington, DE, U.S.A.) at 260 nm and the purity of the RNA was confirmed using the ratio of 260/280 nm. One microgram of total RNA was used for the synthesis of cDNA using the Maxime RT PreMix kit (Intron Biotechnology, Seoul, Korea). Quantitative real-time (qRT)-PCR was performed using a CFX96TM Real-Time PCR Detection System (Bio-Rad) with iQTM SYBR Green Supermix (Bio-Rad) and gene specific primer sets (PHS-001050, Accutarget™ human antioxidant real-time PCR primer set Bioneer, Seoul, Korea).23) The primer sets for Nrf2, HO-1, NQO-1 and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) are listed in Table 1. In brief, samples were heated to 95°C for 5 min followed by 40 cycles each of 95°C (10 s), 42°C (10 s) and 72°C (20 s). Melting curve analysis was performed to ensure the amplification of a single amplicon. Samples were analyzed via the ΔΔCT method using glyceraldehyde 3-phosphate dehydrogenase for normalization. Data are presented as the fold-change from control samples. Each exposure condition for mRNA expression was confirmed by two additional independent experiments, representing biological duplicates.
Table 1. Primer Sets Used in the Study
| Gene | Primer sequences | Amplicon sizes |
|---|
| Nrf2 (F)Nrf2 (R) | 5′-GCGACGGAAAGAGTATGAC-3′5′-TTGGCAGATCCACTGGTTT-3′ | 99 bp |
| HO-1 (F)HO-1 (R) | 5′-GCAACCCGACAGCATGC-3′5′-TGCGGTCGAGCTCTTCTG-3′ | 245 bp |
| NQO1 (F)NQO1 (R) | 5′-CGCAGACCTTGTGATATTCCAG -3′5′-CGTTTCTTCCATCCTTCCAGG -3′ | 249 bp |
| GAPDH (F)GAPDH (R) | 5′-TCCCATCACCATCTTCCA-3′5′-CATCACGCCACAGTTTCC-3′ | 380 bp |
BEAS-2B cells were transiently transfected with control siRNA (sc-37007) and Nrf2 siRNA (sc-37030) mixed with transfection reagent (Santa Cruz Biotechnology) according to manufacturer’s protocol. In brief, cells were seeded in antibiotic-free normal growth medium and incubated at 37°C in CO2 incubator until the cells get 60–80% confluency. For each transfection, 6 µL siRNA duplex and siRNA transfection reagent (sc-29528) were diluted in 100 µL siRNA tranfection medium (sc-36868). After that, siRNA duplex and siRNA transfection reagent were mixed gently by pipetting the solution up and down, and then incubated for 30 min at room temperature. Then cells were washed once with 2 mL transfection medium. The mixture of siRNA duplex and siRNA reagent was then added to the cells with 800 µL transfection medium and then overlayed onto the cells. The cells were incubated at 37°C in CO2 incubator for 6 h. After incubation, the transfection medium was aspired and added normal growth medium for additional 24 h incubation in normal cell culture condition. Finally the cells were used for additional experiment such as western blot and cell viability assay.
Statistical AnalysisThe statistical significant among three independent experiments was analyzed by Student’s t-test and assuming equal variance. All of the results were considered significant if the p value was <0.05. Data was expressed in graphs as mean±S.D.
Results
Protection of BEAS-2B Cells against PQ-Induced Death by LA TreatmentThe present study aimed to determine the potential bronchial protective effect of LA against PQ-induced cell death. BEAS-2B cells were incubated at a range of PQ concentrations for 48 h and cell viability was evaluated by the MTT assay. PQ consistently and dose-dependently induced cell death in BEAS-2B cells (Fig. 1A). After 48 h incubation, cell viability was significantly reduced when the cells were exposed to 0.2–1 mm PQ. The next experiment examined the cytotoxicity of LA on BEAS-2B cells (Fig. 1B). LA concentrations from 0.1–1 mm LA were not appreciably cytotoxic to BEAS-2B cells. Interestingly, LA co-treatment for 48 h recovered viability of PQ-treated cell populations from 60% to 100%. Even 0.3 mm LA achieved recovery of almost 40%. We further evaluated morphological changes of BEAS-2B cells treated with PQ and PQ with LA by light microscopy (Figs. 1D–G). Cells were exposed to 0.2 mm PQ displayed a rounded and shrunken shape (Fig. 1F) and attached less tenaciously as compared to untreated BEAS-2B cells (Fig. 1D). Interestingly, the cells co-treated with PQ and LA (Fig. 1G) were significantly healthier in appearance than PQ-treated cells (Fig. 1F), and display a morphology that was similar to untreated cells (Fig. 1D). Cells treated with 0.2 mm LA likewise displayed a similar morphology as untreated cells. The observations supported the view that LA has a cytoprotective effect on PQ-exposed cells.

To decipher the effect of LA on LDH activity during oxidative stress, we evaluated the effect of LA on LDH enzyme activity in PQ-treated BEAS-2B cells (Fig. 2). PQ increased LDH enzyme activity in concentration- and time-dependent manners, with a loss of cellular integrity (Figs. 2A, B). The release of LDH to the medium was significantly increased at PQ doses of 0.1 mm and higher (Figs. 2A–C), consistent with PQ-mediated damage of cellular membrane integrity. In contrast, co-treatment with LA and PQ condition appreciably reduced LDH release in time- and dose-dependent manner (Figs. 2B, C). LDH activity in the presence of 0.2 mm PQ and 0.5 mm LA was reduced from 59% to 38% at 6 h (Fig. 2B). These data suggested that LA could protect cellular integrity of PQ-treated cells. We then extended the examination of LDH activity to determine the effect of various doses of LA on BEAS-2B cells after a 6-h PQ challenge. Different dose of LA consistently preserved cellular integrity of PQ-treated cells (Fig. 2C).

We tested whether LA had any beneficial effect on total ROS generation in the presence of PQ. Significant ROS generation was observed when cells were exposed to 0.2 mm PQ (Figs. 3A, B). Administration of pyocyanin, which is a potent ROS inducer, dramatically increased total ROS content in BEAS-2B cells compared to control cells (Fig. 3A). However, this increased total ROS generation was not found in cells exposed solely to LA (Fig. 3A). The ROS inhibitor NAC markedly reduced total ROS from approximately 90% to about 40% (Fig. 3A). Interestingly, a marked reduction of total ROS generation was also observed in PQ-treated BEAS-2B cells that were exposed to 0.5 mm LA (Fig. 3A). According to our quantitative data in Fig. 3B, the reduction was greater than observed with NAC treatment. These results supported the role of LA as a potent ROS scavenger in the presence of PQ. Furthermore, to ascertain whether LA was influential on lipid peroxidation induced by PQ, we measured the level of lipid peroxidation using the thiobarbituric acid reactive substances (TBARS) method.24) As shown in Fig. 3C, PQ exposure led to the significant production of MDA levels as compared to normal cells in BEAS-2B cells at 24 h. With 0.5 mm, LA did not influence MDA levels. However, co-treatment with 0.2 mm PQ and 0.5 mm LA significantly abated the levels of MDA. The results indicated that LA suppressed total MDA level in presence of PQ, which caused oxidative stress in the bronchial epithelial cells.

The Nrf2 signaling pathway plays a central regulator in the adaptive response to chemical and oxidative stress, and maintains the cellular defense system. To investigate at the molecular level whether the observed protective effect of LA was conferred by the activation of the Nrf2 signaling pathway in BEAS-2B cells, we tested the expression of Nrf2, HO-1 and NQO1 mRNA in the presence of LA (Fig. 4A). Interestingly, LA itself can induce Nrf2, HO-1 and NQO1 mRNA expression dose dependently. A significant induction of Nrf2 mRNA was observed at concentrations as low as 0.1 mm and reached a 5-fold induction at 0.5 mm at 3 h (Fig. 4A). LA effectively activated the expression of Nrf2 in PQ-treated cells (Fig. 4B). HO-1 and NQO1 are two major elements of cellular defense mechanisms against oxidative stress; they are also regulated by the Nrf2 signaling pathway. Thus, we examined whether LA could induce HO-1 and NQO1 expression in PQ-exposed BEAS-2B cells. To examine the expression of HO-1 and NQO1 mRNA, cells were treated with different doses of PQ and LA for 3 h. Co-treatment with PQ and LA resulted in significant HO-1 and NQO1 mRNA expression accompanied by HO-1 and NQO1 protein expression (Figs. 4C, D). Taken together, this data indicated that LA elicits Nrf2 and its target HO-1 and NQO1 expression in BEAS-2B cells.
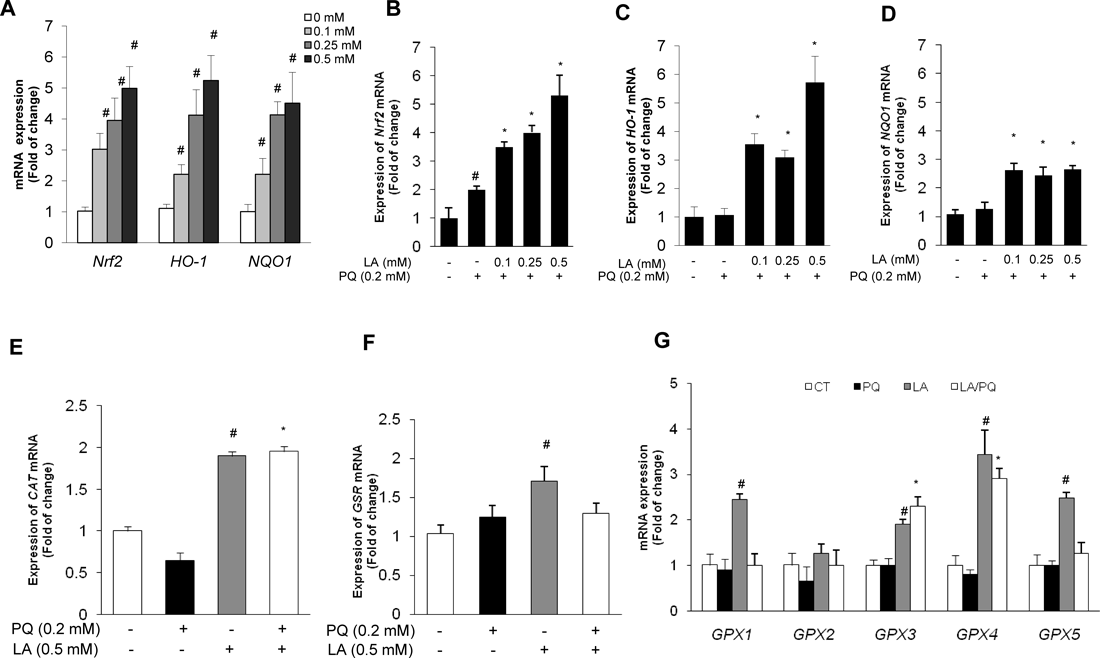
Induction of antioxidant related CAT, GSR, and GPX family genes by LA in BEAS-2B cells. A basal quantification of the expressions of endogenous antioxidant related genes, including CAT, GSR and GPX family were determined in LA-treated BEAS-2B cells in the presence and absence of PQ-induced oxidative stress. To discern the extent of LA-mediated antioxidant capacity in BEAS-2B cells, quantitative RT-PCR analysis was performed to investigate mRNA expression in BEAS-2B cells. We observed a significant induction in CAT but not in GSR mRNA expression in BEAS-2B cells (Figs. 4E, F). PQ exposure led to significant decrease in CAT mRNA. Surprisingly, the same trend was not observed in GSR mRNA expression, while co-incubation with 0.5 mm LA significantly induced CAT mRNA expression more than 2-fold comparing with the control group (Figs. 4E, F). Next, we tested GPX family mRNA expression in the presence and absence of PQ with or without 0.5 mm LA. PQ treatment did not interfere with the expression of the GPX family. A large increase of GPX1, GPX3, GPX4 and GPX5 was noted in the presence of LA, while GPX2 was not induced (Fig. 4G). Interestingly, GPX3 and GPX4 were greatly induced by LA in the PQ-induced condition. GPX1 and GPX5 mRNA expression was induced when cells were treated solely with LA but not in presence of PQ. The data suggested that LA activates the antioxidant system in BEAS-2B cells, indicating that some antioxidant activity can be maintained by LA.
Next, the protein expression of Nrf2 in the presence of LA was examined. BEAS-2B cells were treated with a variety of LA concentrations and Nrf2 expression was detected. LA induced Nrf2 expression with time and dose-dependent manner in BEAS-2B cells (Figs. 5A, B). Co-treatment with LA and 0.2 mm PQ effectively activated Nrf2 and its target HO-1 and NQO1 expression (Figs. 5C, D). However, the maximal induction of Nrf2, HO-1 and NQO1 expression was diminished after 6 h (Fig. 5D). We next examined the response in cellular and nuclear Nrf2 levels during LA treatment in the PQ-exposed condition. Western blot analysis indicated that nuclear Nrf2 protein levels were stabilized by LA in PQ-induced BEAS-2B cells (Fig. 5E). On the other hand, cellular Nrf2 levels declined in co-treatment condition with PQ and LA (Fig. 5E). To further evaluate respective role of Nrf2 in the cellular protection exerted by LA, BEAS-2B cells were transfected with siRNA against both Nrf2 and control. Nrf2 siRNA transfection led to the knockdown of Nrf2 protein to an undetectable level as determined by western blot, whereas control siRNA showed no effect in Nrf2 protein level (Fig. 5F). Nrf2 knockdown cells showed increased susceptibility to PQ-exposure reflected by cell viability assay compared to control-siRNA transfected cells (Fig. 5G). The results suggested that Nrf2 activation is an essential event for LA mediated protection effect against ROS insult.

Discussion
LA is a small natural molecule that is normally present in small quantities in meats and vegetables, where it functions as a biological antioxidant. Its antioxidant activity has been highly implicated in the treatment of various human diseases such as cancer and diabetes.25) Furthermore, LA has a beneficial effect on various oxidative conditions such as lipopolysaccharide-induced oxidative stress, and is able to generate endogenous antioxidant.25,26) However, its effect on PQ intoxication has not been well studied. These facts led to the hypothesis concerning its effect on PQ-exposed BEAS-2B cells. Treatment of 0.5 mm LA impressively improved cell viability and decline in total intracellular ROS generation induced by PQ. This finding is part of the ROS scavenging effect by LA on PQ-exposed BEAS-2B cells. In addition, it is possible that the observed decline of ROS may be associated with the high reactivity of the hydroxyl group of LA, which reacts with free radicals and converts them to more benign molecules.27)
A number of earlier studies have demonstrated that Nrf2 protects various cell types including lung, liver, kidney, brain and intestine against environmental insults.23,28,29) The present study provides evidence that LA confers cell protection through the activation of Nrf2 and its down-stream target genes HO-1 and NQO1, and some other antioxidant related genes. He et al. demonstrated that PQ can activate Nrf2 expression in various cell lines.30) However, the induction of Nrf2 mRNA by PQ is transient. Furthermore, this adaptive response of Nrf2 to ROS stress induced by PQ is not enough to activate Nrf2 target gene expression. Induction of Nrf2 mRNA was 2-fold increased by PQ only at 3 h but the induction of Nrf2 target genes, HO-1 and NQO1 was not detected. However, the induction of Nrf2 by LA treatment is different mechanism. Although Nrf2 half-life is short (less than 20 min), 0.5 mm LA treatment can induce Nrf2 persistently up to 48 h. Persistent induction of Nrf2 mRNA in oxidative condition is required for adequate cellular protection. Induction of phase II enzymes that plays as indirect antioxidants via neutralizing reactive electrophiles is regulated by Nrf2. Therefore, the effective induction of Nrf2 by LA treatment leads its nuclear accumulation and transactivation which can enhance de novo synthesis of antioxidant-related genes. In addition, Kwak et al. revealed that Nrf2 expression can auto-regulate through its own ARE-like sequences on the promoter and lead sustained phase II gene expression.31) Therefore, we can speculate that LA can induce Nrf2 de novo synthesis via its autoregulation.
Cellular stability of Nrf2 and its nuclear translocation are important for proper cytoprotective function against oxidative stresses. An earlier study identified two captious residues named C273 and C288 on Keap1 that are involved in Nrf2 ubiquitination. The ubiquitination mechanisms are likely responsible for the rapid Nrf2 disassociation from Keap1 and short half-life of Nrf2. This finding was supported by the Rubio et al., where Nrf2 was induced early in BEAS-2B cells.6) A recent study demonstrated that Nrf2-dependent induction of 20S proteasome and Pa28αβ (11S) is also important for cellular defense against oxidative stress.32) In present study, we also observed that LA can facilitate nuclear translocation of Nrf2. We can speculate that LA might activate Nrf2 through both the dissociation of Keap1 and the phosphorylation of Nrf2 itself, although the mechanisms need further investigation.
In addition, many studies have reported that Nrf2 can regulate phage II enzyme/antioxidant genes such as HO-1, NQO1, CAT and GSR via ARE binding site on the promoter region of the target genes. HO-1 and NQO1, in particular, have emerged as important mediators of cellular defense against wide-ranging tissue injuries and have been suggested to be therapeutic targets in various disease models.9,12) These elements accommodate ARE sites in their promoter region, and are subjected to response by the master transcription factor Nrf2.9,12) Indeed, in our bronchial epithelium cell system, LA treatment can induce HO-1 and NQO1 expression with a dose-dependent manner. An another important way of counteracting PQ-induced toxicities in the human bronchus may be by enhancing the level of other antioxidant genes like CAT, GSR and the GPX family.33) Our results showed that LA could incite ARE-independent CAT mRNA expression but failed to influence Nrf2-dependent GSR mRNA expression. We have also quantified a group of antioxidant GPX mRNA expression. The expression of GPXs, a typical class of antioxidant enzymes, increases during oxidative stress, which can remove ROS effectively and reduce damage caused by oxidative stress.34) It has been elucidated that GPX1 is associated with attenuation of NADPH and NADH oxidation, and its expression is implicated in protection against moderate oxidative stress induced by PQ in mice.35,36) GPX2 and GPX3 genes are members of the glutathione peroxidase family encoding a selenium-dependent glutathione peroxidase. GPX3 can give protection against oxidative damage via catalyzing the reduction of hydrogen peroxide, lipid peroxides and organic hydroperoxide, by glutathione while the function of GPX2 has not been well elucidated in oxidative stress. A phospholipid hydroperoxidase, GPX4 can protect cells against membrane lipid peroxidation.37) It has been demonstrated that GPX5 is a selenium-independent gene, and it participates in cell protection by interfering lipid peroxidation level and/or by ROS scavenging effect.38)
In this study, only GPX3 and GPX4 were induced by LA in PQ exposed BEAS-2B cells. Up-regulation of GPX4 expression by LA may contribute to cell protection by reducing lipid peroxidation. LA significantly induced the expression of GPX1 and GPX5, but LA notably failed to induce in the PQ condition. Interestingly, GPX2 was not detected in the same condition. We speculate that these altered expressions of antioxidant genes are controlled by alternative transcription pathways governed by NF-κB or AP-1, rather than Nrf2. Of course, this prospect demands further investigation.
Membrane lipids are certainly susceptible to oxidation due to their high concentration of polyunsaturated fatty acids and their association with enzymatic and non-enzymatic events in cells. The generated free radicals react with lipids, causing peroxidation and resulting in release of products such as MDA, hydroperoxides and hydroxyl radicals. Peroxidation of lipids causes membrane damages and disruption.39) In this study, we observed that LA significantly reduced PQ-mediated MDA level. The result supports previous observations.40) The reduction in the level of MDA was ascribed to the potent free radical scavenging capacity of LA. As thiols play a significant role in sheltering cells against lipid peroxidation, it was postulated that the dithiol nature of LA is responsible for the suppression of lipid peroxidation levels.41) Other feasible interventions of LA preclude scavenging of fatty acid peroxyl radicals occurring during lipoperoxidative processes and inhibition of 15-lipoxigenase oxidative activity.42) The present data support the thought that LA could target these plausible mechanisms.
In summary, the natural compound LA can protect human bronchial epithelial cells from PQ-induced ROS stress. This protective effect is mediated by the stabilization of Nrf2 and induction of its downstream target genes HO-1 and NQO1, as well as other cellular antioxidant genes including CAT. These findings shed light on the mechanisms of the protection afforded by LA against PQ-induced oxidative stress.
Acknowledgment
Authors thank to Mrs. Tamanna Zerin for her technical supports and valuable comments. This work was supported by the Soonchunhyang University Research Fund (No. 20110697) for Y.-S. Kim.
REFERENCES
- 1) Suntres ZE, Omri A, Shek PN. Pseudomonas aeruginosa-induced lung injury: role of oxidative stress. Microb. Pathog., 32, 27–34 (2002).
- 2) Smith LL, Lewis CP, Wyatt I, Cohen GM. The importance of epithelial uptake systems in lung toxicity. Environ. Health Perspect., 85, 25–30 (1990).
- 3) Fuentes-Mattei E, Rivera E, Gioda A, Sanchez-Rivera D, Roman-Velazquez FR, Jimenez-Velez BD. Use of human bronchial epithelial cells (BEAS-2B) to study immunological markers resulting from exposure to PM(2.5) organic extract from Puerto Rico. Toxicol. Appl. Pharmacol., 243, 381–389 (2010).
- 4) Kinnula VL, Yankaskas JR, Chang L, Virtanen I, Linnala A, Kang BH, Crapo JD. Primary and immortalized (BEAS 2B) human bronchial epithelial cells have significant antioxidative capacity in vitro. Am. J. Respir. Cell Mol. Biol., 11, 568–576 (1994).
- 5) Li N, Alam J, Venkatesan MI, Eiguren-Fernandez A, Schmitz D, Di Stefano E, Slaughter N, Killeen E, Wang X, Huang A, Wang M, Miguel AH, Cho A, Sioutas C, Nel AE. Nrf2 is a key transcription factor that regulates antioxidant defense in macrophages and epithelial cells: protecting against the proinflammatory and oxidizing effects of diesel exhaust chemicals. J. Immunol., 173, 3467–3481 (2004).
- 6) Rubio V, Zhang J, Valverde M, Rojas E, Shi ZZ. Essential role of Nrf2 in protection against hydroquinone- and benzoquinone-induced cytotoxicity. Toxicol. In Vitro, 25, 521–529 (2011).
- 7) Bus JS, Gibson JE. Paraquat: model for oxidant-initiated toxicity. Environ. Health Perspect., 55, 37–46 (1984).
- 8) Carré P, Léophonte P. Cytokines and pulmonary fibroses. Rev. Mal. Respir., 10, 193–207 (1993).
- 9) Surh YJ, Kundu JK, Na HK. Nrf2 as a master redox switch in turning on the cellular signaling involved in the induction of cytoprotective genes by some chemopreventive phytochemicals. Planta Med., 74, 1526–1539 (2008).
- 10) Chen XL, Dodd G, Thomas S, Zhang X, Wasserman MA, Rovin BH, Kunsch C. Activation of Nrf2/ARE pathway protects endothelial cells from oxidant injury and inhibits inflammatory gene expression. Am. J. Physiol. Heart Circ. Physiol., 290, H1862–H1870 (2006).
- 11) Thimmulappa RK, Mai KH, Srisuma S, Kensler TW, Yamamoto M, Biswal S. Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer Res., 62, 5196–5203 (2002).
- 12) Cho HY, Kleeberger SR. Nrf2 protects against airway disorders. Toxicol. Appl. Pharmacol., 244, 43–56 (2010).
- 13) Maines MD, Gibbs PE. 30 some years of heme oxygenase: from a “molecular wrecking ball” to a “mesmerizing” trigger of cellular events. Biochem. Biophys. Res. Commun., 338, 568–577 (2005).
- 14) Morse D, Choi AM. Heme oxygenase-1: from bench to bedside. Am. J. Respir. Crit. Care Med., 172, 660–670 (2005).
- 15) Otterbein LE, Lee PJ, Chin BY, Petrache I, Camhi SL, Alam J, Choi AM. Protective effects of heme oxygenase-1 in acute lung injury. Chest, 116 (Suppl.), 61S–63S (1999).
- 16) Nioi P, Hayes JD. Contribution of NAD(P)H:quinone oxidoreductase 1 to protection against carcinogenesis, and regulation of its gene by the Nrf2 basic-region leucine zipper and the arylhydrocarbon receptor basic helix-loop-helix transcription factors. Mutat. Res., 555, 149–171 (2004).
- 17) Dudek M, Bednarski M, Bilska A, Iciek M, Sokołowska-Jezewicz M, Filipek B, Włodek L. The role of lipoic acid in prevention of nitroglycerin tolerance. Eur. J. Pharmacol., 591, 203–210 (2008).
- 18) Packer L, Tritschler HJ, Wessel K. Neuroprotection by the metabolic antioxidant alpha-lipoic acid. Free Radic. Biol. Med., 22, 359–378 (1997).
- 19) Bustamante J, Lodge JK, Marcocci L, Tritschler HJ, Packer L, Rihn BH. Alpha-lipoic acid in liver metabolism and disease. Free Radic. Biol. Med., 24, 1023–1039 (1998).
- 20) Kagan VE, Shvedova A, Serbinova E, Khan S, Swanson C, Powell R, Packer L. Dihydrolipoic acid—a universal antioxidant both in the membrane and in the aqueous phase. Reduction of peroxyl, ascorbyl and chromanoxyl radicals. Biochem. Pharmacol., 44, 1637–1649 (1992).
- 21) Cadirci E, Altunkaynak BZ, Halici Z, Odabasoglu F, Uyanik MH, Gundogdu C, Suleyman H, Halici M, Albayrak M, Unal B. Alpha-lipoic acid as a potential target for the treatment of lung injury caused by cecal ligation and puncture-induced sepsis model in rats. Shock, 33, 479–484 (2010).
- 22) Liu R, Ahmed KM, Nantajit D, Rosenthal FS, Hai CX, Li JJ. Therapeutic effects of alpha-lipoic acid on bleomycin-induced pulmonary fibrosis in rats. Int. J. Mol. Med., 19, 865–873 (2007).
- 23) Zerin T, Kim YS, Hong SY, Song HY. Quercetin reduces oxidative damage induced by paraquat via modulating expression of antioxidant genes in A549 cells. J. Appl. Toxicol. (2012) [Epub on PubMed].
- 24) Ogasawara Y, Ishii K. Exposure to chrysotile asbestos causes carbonylation of glucose 6-phosphate dehydrogenase through a reaction with lipid peroxidation products in human lung epithelial cells. Toxicol. Lett., 195, 1–8 (2010).
- 25) Gorąca A, Huk-Kolega H, Piechota A, Kleniewska P, Ciejka E, Skibska B. Lipoic acid - biological activity and therapeutic potential. Pharmacol. Rep., 63, 849–858 (2011).
- 26) Skibska B, Józefowicz-Okonkwo G, Goraca A. Protective effects of early administration of alpha-lipoic acid against lipopolysaccharide-induced plasma lipid peroxidation. Pharmacol. Rep., 58, 399–404 (2006).
- 27) Bitar MS, Ayed AK, Abdel-Halim SM, Isenovic ER, Al-Mulla F. Inflammation and apoptosis in aortic tissues of aged type II diabetes: amelioration with alpha-lipoic acid through phosphatidylinositol 3-kinase/Akt- dependent mechanism. Life Sci., 86, 844–853 (2010).
- 28) Lee JM, Li J, Johnson DA, Stein TD, Kraft AD, Calkins MJ, Jakel RJ, Johnson JA. Nrf2, a multi-organ protector? FASEB J., 19, 1061–1066 (2005).
- 29) Moi P, Chan K, Asunis I, Cao A, Kan YW. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc. Natl. Acad. Sci. U.S.A., 91, 9926–9930 (1994).
- 30) He X, Lin GX, Chen MG, Zhang JX, Ma Q. Protection against chromium (VI)-induced oxidative stress and apoptosis by Nrf2. Recruiting Nrf2 into the nucleus and disrupting the nuclear Nrf2/Keap1 association. Toxicol. Sci., 98, 298–309 (2007).
- 31) Kwak MK, Itoh K, Yamamoto M, Kensler TW. Enhanced expression of the transcription factor Nrf2 by cancer chemopreventive agents: role of antioxidant response element-like sequences in the nrf2 promoter. Mol. Cell. Biol., 22, 2883–2892 (2002).
- 32) Pickering AM, Linder RA, Zhang H, Forman HJ, Davies KJ. Nrf2-dependent induction of proteasome and Pa28αβ regulator are required for adaptation to oxidative stress. J. Biol. Chem., 287, 10021–10031 (2012).
- 33) Podder B, Kim YS, Zerin T, Song HY. Antioxidant effect of silymarin on paraquat-induced human lung adenocarcinoma A549 cell line. Food Chem. Toxicol., 50, 3206–3214 (2012).
- 34) Lubos E, Loscalzo J, Handy DE. Glutathione peroxidase-1 in health and disease: from molecular mechanisms to therapeutic opportunities. Antioxid. Redox Signal., 15, 1957–1997 (2011).
- 35) Lei XG. Glutathione peroxidase-1 gene knockout on body antioxidant defense in mice. Biofactors, 14, 93–99 (2001).
- 36) de Haan JB, Bladier C, Griffiths P, Kelner M, O’Shea RD, Cheung NS, Bronson RT, Silvestro MJ, Wild S, Zheng SS, Beart PM, Hertzog PJ, Kola I. Mice with a homozygous null mutation for the most abundant glutathione peroxidase, Gpx1, show increased susceptibility to the oxidative stress-inducing agents paraquat and hydrogen peroxide. J. Biol. Chem., 273, 22528–22536 (1998).
- 37) Scimeca MS, Lisk DJ, Prolla T, Lei XG. Effects of gpx4 haploid insufficiency on GPx4 activity, selenium concentration, and paraquat-induced protein oxidation in murine tissues. Exp. Biol. Med. (Maywood), 230, 709–714 (2005).
- 38) Noblanc A, Kocer A, Chabory E, Vernet P, Saez F, Cadet R, Conrad M, Drevet JR. Glutathione peroxidases at work on epididymal spermatozoa: an example of the dual effect of reactive oxygen species on mammalian male fertilizing ability. J. Androl., 32, 641–650 (2011).
- 39) Kim HS, Kwack SJ, Lee BM. Lipid peroxidation, antioxidant enzymes, and benzo[a]pyrene-quinones in the blood of rats treated with benzo[a]pyrene. Chem. Biol. Interact., 127, 139–150 (2000).
- 40) Dadhania VP, Tripathi DN, Vikram A, Ramarao P, Jena GB. Intervention of alpha-lipoic acid ameliorates methotrexate-induced oxidative stress and genotoxicity: A study in rat intestine. Chem. Biol. Interact., 183, 85–97 (2010).
- 41) Jesudason EP, Masilamoni JG, Jebaraj CE, Paul SF, Jayakumar R. Efficacy of dl-alpha lipoic acid against systemic inflammation-induced mice: antioxidant defense system. Mol. Cell. Biochem., 313, 113–123 (2008).
- 42) Lapenna D, Ciofani G, Pierdomenico SD, Giamberardino MA, Cuccurullo F. Dihydrolipoic acid inhibits 15-lipoxygenase-dependent lipid peroxidation. Free Radic. Biol. Med., 35, 1203–1209 (2003).