Abstract
Our recent work has shown that cannabidiol (CBD) exhibits the most potent direct inhibition of human cytochrome P450 1A1 (CYP1A1) among the CYP enzymes examined. However, the mechanism underlying this CBD inhibition remains to be clarified. Thus, to elucidate the structural requirements for the potent inhibition by CBD, the effects of CBD and its structurally related compounds on CYP1A1 activity were investigated with recombinant human CYP1A1. Olivetol, which corresponds to the pentylresorcinol moiety of CBD, inhibited the 7-ethoxyresorufin O-deethylase activity of CYP1A1; its inhibitory effect (IC50=13.8 µM) was less potent than that of CBD (IC50=0.355 µM). In contrast, d-limonene, which corresponds to the terpene moiety of CBD, only slightly inhibited CYP1A1 activity. CBD-2′-monomethyl ether (CBDM) and CBD-2′,6′-dimethyl ether inhibited CYP1A1 activity with IC50 values of 4.07 and 23.0 µM, respectively, indicating that their inhibitory effects attenuated depending on the level of methylation on the free phenolic hydroxyl groups in the pentylresorcinol moiety of CBD. Cannabidivarin inhibited CYP1A1 activity, although its inhibitory potency (IC50=1.85 µM) was lower than that of CBD. The inhibitory effects of Δ9-tetrahydrocannabinol and cannabielsoin (IC50s ≈10 µM), which contain a free phenolic hydroxyl group and are structurally constrained, were less potent than that of CBDM, which contains a free phenolic hydroxyl group and is rotatable between pentylresorcinol and terpene moieties. These results suggest that the pentylresorcinol structure in CBD may have structurally important roles in direct CYP1A1 inhibition, although the whole structure of CBD is required for overall inhibition.
Cannabidiol (CBD), one of the major constituents in marijuana,1) is not psychoactive but has several pharmacological effects such as prolonging drug-induced sleep, antiepileptic, anxiolytic, and antiemetic actions.2) CBD is also known to inhibit hepatic microsomal drug metabolism in mammals.3–5) A previous clinical study has shown that the administration of CBD reduces the systemic clearance of hexobarbital, which is metabolized by cytochrome P450 2C9 (CYP2C9), in human subjects.6) In vitro studies by Jaeger et al.7) and us8–13) demonstrated that CBD inhibits catalytic activities of CYP1A1, CYP1A2, CYP2B6, CYP2C9, CYP2C19, CYP2D6, CYP3A4, and CYP3A5 in human liver microsomes and/or recombinant CYP enzymes. Interestingly, we have recently reported that the CBD-mediated direct inhibition of human CYP1A1 activity is the strongest among those of CYP activities reported so far.8) In addition, CYP1A1 was more potently inhibited by CBD than by the other major phytocannabinoids examined.8)
CYP1A1 is expressed in various tissues including the liver and lung,14–16) although the constitutive expression level is very low. In general, hepatic and pulmonary CYP1A1 is induced by exposure to tobacco smoke.17,18) CYP1A1 is important in the bioactivation of procarcinogens, such as polycyclic aromatic hydrocarbons and heterocyclic amines.16,19) Furthermore, this enzyme plays a role in the metabolism of several drugs including granisetron20) and dacarbazine.21) Therefore, potent inhibition of CYP1A1 by CBD may not only lead to the suppression of metabolic activation of procarcinogens but also influence the pharmacokinetics of drugs metabolized mainly by CYP1A1. However, it is not clear which moiety of CBD contributes to potent direct CYP1A1 inhibition.
CBD contains pentylresorcinol and terpene moieties and is rotatable between their moieties (Fig. 1). Our previous study indicated that two free phenolic hydroxyl groups in the pentylresorcinol moiety of CBD could contribute to the inhibitory effects of the phytocannabinoid on p-nitroanisole O-demethylation and aniline hydroxylation by mouse liver microsomes.22) Recently, we have elucidated the structural requirements for inhibition of cyclooxygenase, lipoxygenase, and CYP by CBD and its derivatives.9–11,23,24) These studies on structure–inhibition relationships could provide very useful information to account for the mechanism(s) underlying the potent inhibition of human CYP1A1 by CBD.

Fig. 1. Chemical Structures of CBD-Related Compounds Used in This Study
In the present study, we investigated the inhibitory effects of compounds structurally related to CBD (Fig. 1) on human CYP1A1 activity to clarify the structural requirements for the potent direct inhibition by CBD. Our study suggests that the pentylresorcinol structure in CBD may play important roles in direct CYP1A1 inhibition, as partly supported by molecular modeling of CYP1A1 with the phytocannabinoid. The underlying mechanism is discussed in this paper.
Materials and Methods
MaterialsCBD and Δ9-tetrahydrocannabinol (Δ9-THC) were isolated from cannabis leaves using a method previously reported.25) CBD-2′-monomethyl ether (CBDM), CBD-2′,6′-dimethyl ether (CBDD), and cannabielsoin (CBE) were prepared as described previously.26) The purities of these cannabinoids were determined to be above 97% by gas chromatography, except for CBDD whose purity was 93%.27) Cannabidivarin (CBDV) was generously provided from Dr. Yukihiro Shoyama at Nagasaki International University (Sasebo, Japan). Other chemicals were obtained from the following sources: microsomes from baculovirus-infected insect cells expressing CYP1A1 with reduced nicotinamide adenine dinucleotide phosphate (NADPH)-CYP reductase (Supersomes™) from BD Gentest (Woburn, MA, U.S.A.); NADPH from Oriental Yeast Co., Ltd. (Tokyo, Japan); 7-ethoxyresorufin, resorufin, olivetol, and d-limonene from Sigma-Aldrich Corp. (St. Louis, MO, U.S.A.); resorcinol from Wako Pure Chemical Industries, Ltd. (Osaka, Japan); orcinol from Tokyo Chemical Industry Co., Ltd. (Tokyo, Japan). All other chemicals and solvents used were of the highest quality commercially available.
Direct Inhibition StudiesThe activity of 7-ethoxyresorufin O-deethylase (EROD) was determined by using a 96-well microtiter plate as reported previously,8) with a minor modification. Briefly, an incubation mixture consisted of recombinant human CYP1A1 (10 fmol), CBD or its structurally related compounds (up to 100 µM), 7-ethoxyresorufin (150 nM), NADPH (1.67 mM), and Tris–HCl buffer (50 mM, pH 7.4) containing 1% fatty acid-free bovine serum albumin in a final volume of 200 µL. After pre-warming at 37°C for 5 min, reactions were initiated by the addition of NADPH. Fluorescence derived from resorufin formation was recorded every 5 min for 30 min using FLUOstar OPTIMA® (BMG Labtech, Offenburg, Germany) with excitation and emission filters at 544 and 590 nm, respectively. We confirmed that the formation of resorufin increased linearly with an incubation time up to 30 min in the presence or absence of CBD-related compounds. The EROD activity was measured after a 10-min incubation in order to minimize the contribution of CBD-related compounds to metabolism-dependent inhibition. The IC50 value was calculated by nonlinear least-squares regression analysis with Origin 7.5J software (OriginLab, Northampton, MA, U.S.A.).
The effects of three or four different inhibitor concentrations on the formation of resorufin were examined at five substrate concentrations to characterize the enzyme kinetics for the inhibition of human CYP1A1 by CBD and its related compounds. The apparent Ki value (inhibition constant) was determined from the x-intercept of a plot of apparent Km/Vmax (obtained from the slope of the Lineweaver–Burk plots) versus inhibitor concentration. The x-intercept, which is equal to −Ki, was calculated by linear regression using Origin 7.5J software (OriginLab). Lineweaver–Burk plots of enzyme kinetic data were generated to determine the mode of inhibition.
Molecular ModelingThe conformation of human CYP1A1 was automatically constructed by Swiss-Model (http://swissmodel.expasy.org/) using the crystallographic data of human CYP1A2 (2HI4) obtained from the Protein Data Bank (http://www.rcsb.org/pdb/). Six peptides of CYP1A1 (Arg-106 to Gly-128, Leu-217 to Gly-229, Lys-252 to Glu-256, Asn-309 to Val-322, Ser-379 to His-388, Met-490 to Lys-499) were extracted as substrate recognition sites. Energy optimization of the model was carried out using Insight II/Discover as described previously.28) The amino acid residues at the active site of CYP1A1 were drawn using RasMol version 2.6-ucb-1.0 as reported previously.28)
Results
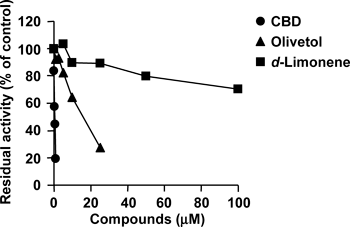
Effects of CBD-Related Compounds on Human CYP1A1 ActivityTo elucidate which moiety of CBD contributes to human CYP1A1 inhibition, the effects of olivetol and d-limonene were examined. Olivetol, which corresponds to the pentylresorcinol moiety of CBD, inhibited the EROD activity of CYP1A1 in a concentration-dependent manner (Fig. 2). The IC50 value of olivetol was 13.8 µM and much higher than that of CBD (IC50=0.355 µM). In contrast, d-limonene, which corresponds to the terpene moiety of CBD, showed slight inhibition under the present conditions (Fig. 2).
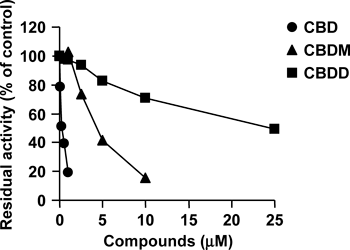
To determine whether free phenolic hydroxyl group(s) in the pentylresorcinol moiety of CBD has a role in CYP1A1 inhibition, the inhibitory effects of two methylated derivatives of CBD were investigated. CYP1A1 activity was inhibited by CBDM and CBDD, which are monomethylated and dimethylated derivatives of CBD, respectively (Fig. 3). The IC50 values of CBDM and CBDD were 4.07 and 23.0 µM, respectively, showing that the extent of the inhibitory effect of CBD on CYP1A1 depended on the number of free phenolic hydroxyl groups in the pentylresorcinol moiety of CBD. This result raised the question whether CBDM and CBDD themselves inhibit CYP1A1 activity or not. If CBDM and CBDD were metabolized by CYP1A1 to form the corresponding O-demethylated metabolites, i.e., CBD and CBDM, respectively, these metabolites might inhibit CYP1A1 activity. Therefore, we examined the formation of O-demethylated metabolites of CBDM and CBDD. When recombinant human CYP1A1 (100 fmol) was incubated with CBDM or CBDD at a substrate concentration of 50 µM for 30 min, the corresponding O-demethylated metabolites were not detected as determined by gas chromatography (data not shown). These results indicated that CBDM and CBDD themselves inhibited CYP1A1 activity.
CBD and olivetol contain a pentyl side chain in their structures. To determine the importance of the pentyl group of CBD in CYP1A1 inhibition, inhibitory potencies of CBDV, orcinol, and resorcinol were examined. CBDV possessing a shorter side chain (i.e., propyl group) inhibited CYP1A1 activity with the IC50 value of 1.85 µM (Fig. 4A). Orcinol, which includes a methyl group in place of the pentyl group in olivetol, concentration-dependently inhibited CYP1A1 activity (Fig. 4B), although its inhibitory effect (IC50=62.9 µM) was less potent than that of olivetol (IC50=13.8 µM). In contrast, resorcinol, which lacks the alkyl side chain of olivetol, did not exert any inhibitory effect (Fig. 4B).

As shown in Fig. 1, CBD is freely rotatable between pentylresorcinol and terpene moieties. To determine what configuration of CBD contributes to CYP1A1 inhibition, inhibition studies were conducted with Δ9-THC and CBE. Δ9-THC and CBE contain a free phenolic hydroxyl group and are structurally constrained because either of the two phenolic hydroxyl groups in the resorcinol moiety is utilized for ring formation with the neighboring carbon atoms at the 2- and 8-positions, respectively, of CBD. Δ9-THC and CBE equivalently inhibited CYP1A1 activity with IC50 values of 10.3 and 9.93 µM, respectively (Fig. 5), although their inhibitory effects were less potent than that of CBDM (IC50=4.07 µM), which contains a free phenolic hydroxyl group and is rotatable between pentylresorcinol and terpene moieties.
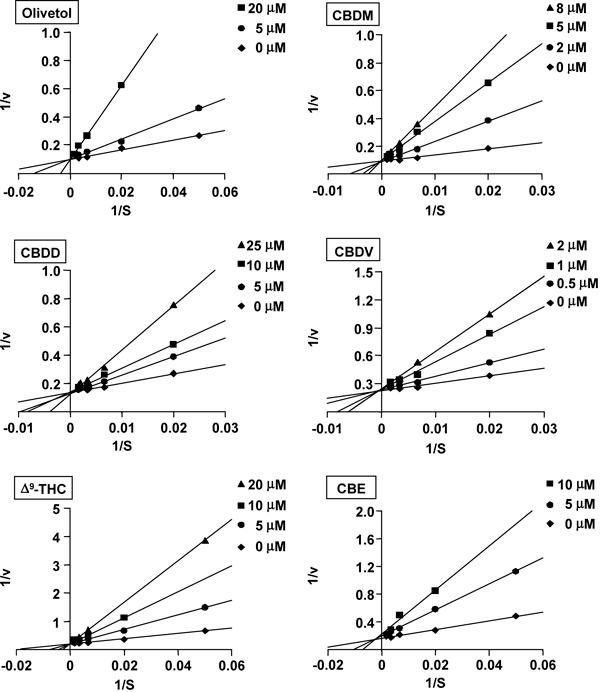
Kinetic Analysis for Direct CYP1A1 Inhibition by Structurally Related Compounds of CBDKinetic analysis for inhibition was conducted to characterize the mode of inhibition of human CYP1A1 activity by the six CBD-related compounds showing inhibition with IC50 values lower than 30 µM. Olivetol, CBDM, CBDD, CBDV, and Δ9-THC, as well as CBD,8) competitively inhibited CYP1A1 activity (Fig. 6). On the other hand, CBE inhibited CYP1A1 activity in a mixed manner (Fig. 6). The apparent Ki values (Table 1) of these compounds correlated well with the corresponding IC50 values (r2=0.980, p<0.0001).
Table 1. Kinetic Parameters for Inhibition of CYP1A1-Mediated EROD Activity by CBD and Its Structurally Related Compounds
| Compounds | r2 a) | Ki (µM) | Mode of inhibition |
|---|
| CBD | 1.00 | 0.155 | Competitive |
| Olivetol | 0.988 | 3.04 | Competitive |
| CBDM | 0.992 | 0.990 | Competitive |
| CBDD | 0.994 | 6.48 | Competitive |
| CBDV | 0.960 | 0.466 | Competitive |
| Δ9-THC | 0.991 | 2.87 | Competitive |
| CBE | 1.00 | 2.49 | Mixed |
a) Correlation coefficient calculated by linear regression of a plot of apparent Km/Vmax versus inhibitor concentration.
Figure 7 shows a model of possible interaction of CBD with human CYP1A1. The phenyl ring of the resorcinol moiety in CBD formed a stacking interaction with Phe-224 in CYP1A1. In addition, two free phenolic hydroxyl groups of CBD were located near a carbonyl oxygen in the side chain of Asn-221 and a carbonyl oxygen in the main chain of Leu-312. The shortest distance from the hydroxyl oxygens of CBD to the carbonyl oxygens of these Asn and Leu residues was 2.53 and 3.72 Å, respectively. Both free phenolic hydroxyl groups of CBD can make hydrogen bonds with these carbonyl oxygens.
Discussion
We have recently reported the potent direct inhibition of human CYP1A1 activity by CBD.8) The competitive inhibition of CYP1A1 by CBD suggested that the phytocannabinoid exerts the inhibitory effect at the active site of CYP1A1.8) This is not surprising because human CYP1A1 is capable of metabolizing CBD.29) On the other hand, it has been shown that CBD is also a mechanism-based inhibitor of CYP1A1.8) However, it is suggested that the contribution of CBD as a mechanism-based inhibitor may be of small importance to CYP1A1 inhibition observed in the present study coincubated CBD with a substrate because the preincubation of a mechanism-based inhibitor with an alternative substrate in the presence of NADPH attenuates inactivation.30,31) In this study, we examined the inhibitory effects of nine CBD-related compounds on CYP1A1 activity to clarify the structural requirements for potent direct CYP1A1 inhibition by CBD under the condition of a short incubation period to minimize the contribution of CBD-related compounds to metabolism-dependent inhibition.
The partial inhibition of CYP1A1 by olivetol but not d-limonene suggests that the pentylresorcinol structure in CBD is essential for inhibition although the whole structure of CBD is required for the overall inhibition of CYP1A1 activity. The structure of the resorcinol moiety in CBD contains two free phenolic hydroxyl groups and a phenyl ring, which are likely to interact with amino acids within the active site of CYP1A1. Inhibition studies with CBDM and CBDD indicate that both free phenolic hydroxyl groups in CBD are required for potent CYP1A1 inhibition. Furthermore, our molecular modeling supports the importance of two free phenolic hydroxyl groups in CYP1A1 inhibition. We have recently reported that CYP2B6, CYP2D6, CYP3A4, and CYP3A5 are more potently inhibited by CBD than by Δ9-THC and cannabinol.9–11) The structural requirements for the inhibitory effects of CBD on these CYP enzymes indicate that both free phenolic hydroxyl groups in the phytocannabinoid contribute to these inhibitions. A similar observation was seen in the inhibitory effects of CBD on CYP-mediated drug oxidations in mouse liver microsomes.22) Thus, these findings suggest that these two free phenolic hydroxyl groups of CBD may be key functional groups common to the inhibition of many CYP enzymes by CBD.
It should be noted that CBDD preserves some degree of the ability to inhibit CYP1A1 activity even though both phenolic hydroxyl groups are methylated. This result reveals that the mechanism underlying CBD-mediated CYP1A1 inhibition cannot be explained only by the presence of two free phenolic hydroxyl groups of the phytocannabinoid. The molecular modeling of CYP1A1 with CBD suggested a possible stacking interaction between the phenyl ring of the resorcinol moiety in CBD and Phe-224 in CYP1A1. The importance of the Phe residue in ligand binding has been shown by previous studies. Lewis et al.32) reported that Ala substitution for Phe-224 in CYP1A1 decreases the catalytic efficiency (Vmax/Km) of EROD activity to approximately one third that of wild-type CYP1A1. For CYP1A2, it has been reported that all mutant enzymes to substitute Ile, Thr, or Tyr residues for Phe-226, which corresponds to Phe-224 in CYP1A1, show considerably reduced EROD activity.33) A crystal structure of CYP1A2 bound to α-naphthoflavone, a representative CYP1 inhibitor, shows that Phe-226 as well as Phe-125 forms a stacking interaction with this inhibitor.34) Phe-224 in CYP1A1 is highly conserved among the CYP1 family.35) Thus, it is conceivable that this residue may interact with the phenyl ring of the resorcinol moiety in CBD. In addition, CBD was accommodated in a torsional conformation of the cyclohexenyl ring of the terpene moiety relative to the phenyl ring of the resorcinol moiety within the active site of CYP1A1 (Fig. 7). This finding is compatible with the results of inhibition studies with Δ9-THC, CBE, and CBDM suggesting that the torsional structure of CBD may be preferentially recognized by CYP1A1. It is surmised that such a torsional conformation of CBD is stabilized by two hydrogen bonds in the CYP1A1 active site, as well as the stacking interaction between the phenyl rings of CBD and Phe-224 in CYP1A1.
There are several studies on the inhibition of human CYP1A1 by naturally occurring compounds containing the resorcinol moiety in their structures.30,36–38) Interestingly, the inhibition of human CYP1A1 by oxyresveratrol having two resorcinol moieties (i.e., 2,4,3′,5′-tetrahydroxystilbene) has been shown to be markedly potentiated by the methylation of all four phenolic hydroxyl groups in oxyresveratrol.36,37) This is in contrast to the result obtained in this study. The presence of free phenolic hydroxyl groups in oxyresveratrol is unlikely to be important in the strong inhibition of human CYP1A1. The role of free phenolic hydroxyl groups for CYP1A1 inhibition may be specific for CBD.
In addition, we focused on a role of the pentyl side chain of CBD in CYP1A1 inhibition because it has been shown that human CYP1A1 is capable of oxidizing the carbon atom at the 1″-position of the pentyl side chain in CBD.29) The side-chain shortening of CBD and olivetol attenuated their inhibitory potencies against CYP1A1 activity. Interestingly, orcinol preserves some degree of the ability to inhibit CYP1A1 activity, although the inhibition by orcinol has not been observed with the other CYP enzymes examined.11,13) The lack of the side chain in the pentylresorcinol structure caused a loss in CYP1A1 inhibition. When the carbon atom at the 1″-position of the pentyl group in CBD comes close to the heme iron, the distal portion of the side chain in CBD may interact with the proposed hydrophobic region within the active site of CYP1A1.32) These results suggest that the pentyl side chain of CBD also plays an important role in potent CYP1A1 inhibition.
CYP1A1 is known to be responsible for the metabolism of several clinically used drugs.20,21) It has been previously reported that granisetron 7-hydroxylation, a major metabolic pathway, is almost exclusively catalyzed by CYP1A1 in human liver microsomes.20) The potent inhibition of CYP1A1 by CBD might influence the pharmacokinetics of granisetron. At present, however, the roles of CYP1A1 and CBD-mediated inhibition in drug clearance are limited because of a limited number of drugs metabolized by CYP1A1 and very low constitutive expression of CYP1A1 in the liver. On the other hand, CYP1A1 is suggested to induce genotoxicity and caricinogenicity by the bioactivation of procarcinogens, such as benzo[a]pyrene and other polycyclic aromatic hydrocarbons.39) Therefore, CBD and its structurally related compounds which potently inhibit CYP1A1 activity would be expected as a lead compound in anticancer chemotherapy.
In conclusion, our inhibition study indicated that two free phenolic hydroxyl groups and the pentyl side chain in the pentylresorcinol moiety of CBD play important roles in direct CYP1A1 inhibition although the whole structure of CBD is essential for potent inhibition. Our molecular modeling supported some results of the inhibition study, i.e., the importance of these two free phenolic hydroxyl groups in CYP1A1 inhibition and the torsional conformation of CBD within the CYP1A1 active site. Furthermore, this modeling speculated a possible stacking interaction between the phenyl ring of CBD and Phe-224 in CYP1A1. This study will provide useful information to understand the precise mechanisms underlying the potent inhibition of human CYP1A1 by CBD.
Acknowledgment
This work was supported in part by a Grant-in-Aid for Young Scientists (B) and Grant-in-Aid for Scientific Research (C) from the Ministry of Education, Culture, Sports, Science and Technology of Japan and by the ‘Academic Frontier’ Project for Private Universities from the Ministry of Education, Culture, Sports, Science and Technology of Japan (2005–2009). We thank Dr. Yukihiro Shoyama (Faculty of Pharmaceutical Sciences, Nagasaki International University, Sasebo, Japan) for generously providing CBDV.
REFERENCES
- 1) ElSohly MA, Slade D. Chemical constituents of marijuana: the complex mixture of natural cannabinoids. Life Sci., 78, 539–548 (2005).
- 2) Mechoulam R, Parker LA, Gallily R. Cannabidiol: an overview of some pharmacological aspects. J. Clin. Pharmacol., 42 (Suppl.), 11S–19S (2002).
- 3) Fernandes M, Warning N, Christ W, Hill R. Interactions of several cannabinoids with the hepatic drug metabolizing system. Biochem. Pharmacol., 22, 2981–2987 (1973).
- 4) Bornheim LM, Borys HK, Karler R. Effect of cannabidiol on cytochrome P-450 and hexobarbital sleep time. Biochem. Pharmacol., 30, 503–507 (1981).
- 5) Hamajima K, Watanabe K, Narimatsu S, Tateoka Y, Yamamoto I, Yoshimura H. Sex difference in the effects of Δ9-tetrahydrocannabinol and cannabidiol on pentobarbital-induced sleeping time and hepatic microsomal drug metabolizing enzyme systems in mice. Yakugaku Zasshi, 103, 1289–1297 (1983).
- 6) Benowitz NL, Nguyen TL, Jones RT, Herning RI, Bachman J. Metabolic and psychophysiologic studies of cannabidiol–hexobarbital interaction. Clin. Pharmacol. Ther., 28, 115–120 (1980).
- 7) Jaeger W, Benet LZ, Bornheim LM. Inhibition of cyclosporine and tetrahydrocannabinol metabolism by cannabidiol in mouse and human microsomes. Xenobiotica, 26, 275–284 (1996).
- 8) Yamaori S, Kushihara M, Yamamoto I, Watanabe K. Characterization of major phytocannabinoids, cannabidiol and cannabinol, as isoform-selective and potent inhibitors of human CYP1 enzymes. Biochem. Pharmacol., 79, 1691–1698 (2010).
- 9) Yamaori S, Maeda C, Yamamoto I, Watanabe K. Differential inhibition of human cytochrome P450 2A6 and 2B6 by major phytocannabinoids. Forensic Toxicol., 29, 117–124 (2011).
- 10) Yamaori S, Ebisawa J, Okushima Y, Yamamoto I, Watanabe K. Potent inhibition of human cytochrome P450 3A isoforms by cannabidiol: role of phenolic hydroxyl groups in the resorcinol moiety. Life Sci., 88, 730–736 (2011).
- 11) Yamaori S, Okamoto Y, Yamamoto I, Watanabe K. Cannabidiol, a major phytocannabinoid, as a potent atypical inhibitor for CYP2D6. Drug Metab. Dispos., 39, 2049–2056 (2011).
- 12) Yamaori S, Koeda K, Kushihara M, Hada Y, Yamamoto I, Watanabe K. Comparison in the in vitro inhibitory effects of major phytocannabinoids and polycyclic aromatic hydrocarbons contained in marijuana smoke on cytochrome P450 2C9 activity. Drug Metab. Pharmacokinet., 27, 294–300 (2012).
- 13) Jiang R, Yamaori S, Okamoto Y, Yamamoto I, Watanabe K. Cannabidiol is a potent inhibitor of the catalytic activity of cytochrome P450 2C19. Drug Metab. Pharmacokinet. (2013), in press.
- 14) Wrighton SA, Campanile C, Thomas PE, Maines SL, Watkins PB, Parker G, Mendez-Picon G, Haniu M, Shively JE, Levin W. Identification of a human liver cytochrome P-450 homologous to the major isosafrole-inducible cytochrome P-450 in the rat. Mol. Pharmacol., 29, 405–410 (1986).
- 15) Shimada T, Yun CH, Yamazaki H, Gautier JC, Beaune PH, Guengerich FP. Characterization of human lung microsomal cytochrome P-450 1A1 and its role in the oxidation of chemical carcinogens. Mol. Pharmacol., 41, 856–864 (1992).
- 16) Shimada T, Hayes CL, Yamazaki H, Amin S, Hecht SS, Guengerich FP, Sutter TR. Activation of chemically diverse procarcinogens by human cytochrome P-450 1B1. Cancer Res., 56, 2979–2984 (1996).
- 17) Willey JC, Coy EL, Frampton MW, Torres A, Apostolakos MJ, Hoehn G, Schuermann WH, Thilly WG, Olson DE, Hammersley JR, Crespi CL, Utell MJ. Quantitative RT-PCR measurement of cytochromes p450 1A1, 1B1, and 2B7, microsomal epoxide hydrolase, and NADPH oxidoreductase expression in lung cells of smokers and nonsmokers. Am. J. Respir. Cell Mol. Biol., 17, 114–124 (1997).
- 18) Kim JH, Sherman ME, Curriero FC, Guengerich FP, Strickland PT, Sutter TR. Expression of cytochromes P450 1A1 and 1B1 in human lung from smokers, non-smokers, and ex-smokers. Toxicol. Appl. Pharmacol., 199, 210–219 (2004).
- 19) Shimada T, Fujii-Kuriyama Y. Metabolic activation of polycyclic aromatic hydrocarbons to carcinogens by cytochromes P450 1A1 and 1B1. Cancer Sci., 95, 1–6 (2004).
- 20) Nakamura H, Ariyoshi N, Okada K, Nakasa H, Nakazawa K, Kitada M. CYP1A1 is a major enzyme responsible for the metabolism of granisetron in human liver microsomes. Curr. Drug Metab., 6, 469–480 (2005).
- 21) Reid JM, Kuffel MJ, Miller JK, Rios R, Ames MM. Metabolic activation of dacarbazine by human cytochromes P450: the role of CYP1A1, CYP1A2, and CYP2E1. Clin. Cancer Res., 5, 2192–2197 (1999).
- 22) Watanabe K, Arai M, Narimatsu S, Yamamoto I, Yoshimura H. Self-catalyzed inactivation of cytochrome P-450 during microsomal metabolism of cannabidiol. Biochem. Pharmacol., 36, 3371–3377 (1987).
- 23) Takeda S, Misawa K, Yamamoto I, Watanabe K. Cannabidiolic acid as a selective cyclooxygenase-2 inhibitory component in cannabis. Drug Metab. Dispos., 36, 1917–1921 (2008).
- 24) Takeda S, Usami N, Yamamoto I, Watanabe K. Cannabidiol-2′,6′-dimethyl ether, a cannabidiol derivative, is a highly potent and selective 15-lipoxygenase inhibitor. Drug Metab. Dispos., 37, 1733–1737 (2009).
- 25) Aramaki H, Tomiyasu N, Yoshimura H, Tsukamoto H. Forensic chemical study on marihuana. I. A detection method of the principal constituents by thin-layer and gas chromatographies. Chem. Pharm. Bull., 16, 822–826 (1968).
- 26) Gohda H, Narimatsu S, Yamamoto I, Yoshimura H. In vivo and in vitro metabolism of cannabidiol monomethyl ether and cannabidiol dimethyl ether in the guinea pig: on the formation mechanism of cannabielsoin-type metabolite from cannabidiol. Chem. Pharm. Bull., 38, 1697–1701 (1990).
- 27) Watanabe K, Motoya E, Matsuzawa N, Funahashi T, Kimura T, Matsunaga T, Arizono K, Yamamoto I. Marijuana extracts possess the effects like the endocrine disrupting chemicals. Toxicology, 206, 471–478 (2005).
- 28) Masuda K, Tamagake K, Okuda Y, Torigoe F, Tsuzuki D, Isobe T, Hichiya H, Hanioka N, Yamamoto S, Narimatsu S. Change in enantioselectivity in bufuralol 1″-hydroxylation by the substitution of phenylalanine-120 by alanine in cytochrome P450 2D6. Chirality, 17, 37–43 (2005).
- 29) Jiang R, Yamaori S, Takeda S, Yamamoto I, Watanabe K. Identification of cytochrome P450 enzymes responsible for metabolism of cannabidiol by human liver microsomes. Life Sci., 89, 165–170 (2011).
- 30) Chang TK, Chen J, Lee WB. Differential inhibition and inactivation of human CYP1 enzymes by trans-resveratrol: evidence for mechanism-based inactivation of CYP1A2. J. Pharmacol. Exp. Ther., 299, 874–882 (2001).
- 31) Kartha JS, Yost GS. Mechanism-based inactivation of lung-selective cytochrome P450 CYP2F enzymes. Drug Metab. Dispos., 36, 155–162 (2008).
- 32) Lewis BC, Mackenzie PI, Miners JO. Comparative homology modeling of human cytochrome P4501A1 (CYP1A1) and confirmation of residues involved in 7-ethoxyresorufin O-deethylation by site-directed mutagenesis and enzyme kinetic analysis. Arch. Biochem. Biophys., 468, 58–69 (2007).
- 33) Parikh A, Josephy PD, Guengerich FP. Selection and characterization of human cytochrome P450 1A2 mutants with altered catalytic properties. Biochemistry, 38, 5283–5289 (1999).
- 34) Sansen S, Yano JK, Reynald RL, Schoch GA, Griffin KJ, Stout CD, Johnson EF. Adaptations for the oxidation of polycyclic aromatic hydrocarbons exhibited by the structure of human P450 1A2. J. Biol. Chem., 282, 14348–14355 (2007).
- 35) Lewis DF, Lake BG, Dickins M, Ueng YF, Goldfarb PS. Homology modelling of human CYP1A2 based on the CYP2C5 crystallographic template structure. Xenobiotica, 33, 239–254 (2003).
- 36) Chun YJ, Kim S, Kim D, Lee SK, Guengerich FP. A new selective and potent inhibitor of human cytochrome P450 1B1 and its application to antimutagenesis. Cancer Res., 61, 8164–8170 (2001).
- 37) Chun YJ, Ryu SY, Jeong TC, Kim MY. Mechanism-based inhibition of human cytochrome P450 1A1 by rhapontigenin. Drug Metab. Dispos., 29, 389–393 (2001).
- 38) Chang TK, Chen J, Yeung EY. Effect of Ginkgo biloba extract on procarcinogen-bioactivating human CYP1 enzymes: identification of isorhamnetin, kaempferol, and quercetin as potent inhibitors of CYP1B1. Toxicol. Appl. Pharmacol., 213, 18–26 (2006).
- 39) Shimada T. Xenobiotic-metabolizing enzymes involved in activation and detoxification of carcinogenic polycyclic aromatic hydrocarbons. Drug Metab. Pharmacokinet., 21, 257–276 (2006).