Abstract
Histamine H1-receptor blockers are used to treat all types of itch resulting from serious skin diseases such as atopic dermatitis, as well as from renal and liver diseases. However, they often lack efficacy in chronic itch, a profound clinical problem that decreases quality of life. The development of effective treatments requires a full understanding of the fundamental mechanisms of itch. Recent studies have indicated that the pathogenic mechanisms of itch also involve agonists other than histamine, including proteases, neuropeptides, cytokines, and opioids, as well as their cognate receptors. Release of these pruritogenic mediators and modulators into the periphery may directly activate itch-mediating C-fibers via specific receptors on the nerve terminals. Histological observations have shown increased epidermal nerve densities in patients with atopic dermatitis, suggesting that the higher density is at least partly responsible for itch sensitization. This hyperinnervation is likely induced by an imbalance between nerve elongation and repulsion factors produced by keratinocytes. Neuronal matrix metalloproteinases are also involved in the penetration of nerve fibers into the extracellular matrix. Moreover, itch-mediating fibers such as gastrin-releasing peptide+ (GRP+) and Mas-related G-protein coupled receptor A3+ (MrgprA3+) fibers are present in the skin. Clinically, emollients or UV-based therapies can partly control epidermal nerve density, but new substances and classes of antipruritic drugs are needed. This review highlights recent knowledge regarding epidermal nerve fibers that are partly involved in itch sensitization, and discuss peripheral mechanisms and treatments of itch, especially in atopic dermatitis.
1. INTRODUCTION
Itch (or pruritus) has been defined as an unpleasant sensation that provokes the desire to scratch. Itch is also believed to signal danger from various environmental factors or physiological abnormalities. Clinically, chronic itch is a burdensome clinical problem that decreases quality of life,1) and it frequently accompanies a variety of inflammatory skin conditions and systemic diseases. Recent studies have indicated that chronic itch is associated with increases in insomnia2) and suicide3) and reductions in patient productivity at work and in the classroom.4) The development of anti-pruritic treatments therefore requires an understanding of the fundamental mechanisms of itch.
Itch and pain are two basic modalities that are initiated and mediated by primary sensory neurons with cell bodies in the dorsal root ganglia (DRG) and trigeminal ganglia. These neurons are highly diverse in somal sizes, expression of ion channels and receptors, innervation territories, and electrophysiological properties.5) Small-diameter DRG neurons with unmyelinated axons (C-fibers) are the major neuronal types that mediate itch and pain.5,6) The sensations of itch and pain are distinct, and each can elicit different behavioral responses, such as scratching to remove irritants and withdrawal to avoid tissue injury, respectively.
Recent studies have implicated histamine-dependent and histamine-independent pathways in transmitting itch. Other systems, including proteases, neuropeptides, cytokines, and opioids, and their cognate receptors, such as thermoreceptors, proteinase-activated receptors (PARs), Mas-related G-protein coupled receptor (Mrgprs) and opioid receptors, are involved in the histamine-independent itch pathway. These pruritogenic mediators and modulators, released in the periphery, may directly activate itch-mediating fibers, especially C-fibers, by binding to specific receptors on the nerve terminals.5,7) These systems also cross-sensitize each other in the enhancement of itch.8) In addition, cutaneous nerve fibers are activated by exogenous mechanical, chemical, and biological stimuli, resulting in itch responses.7,9) Therefore, they are regarded as anti-pruritic targets in patients with chronic itch, including those with atopic dermatitis.
2. HISTOLOGY OF CUTANEOUS NERVE FIBERS IN ATOPIC DERMATITIS
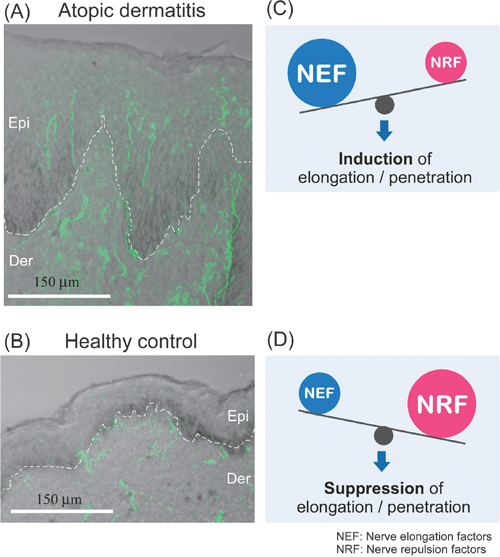
Histological investigations have shown that the density of epidermal nerve fibers is higher in the skin of patients with atopic dermatitis (Fig. 1A) and xerosis than in healthy controls5,7) (Fig. 1B), although the increases of nerve density in patients with pruigo nodularis and psoriasis remain unclear.10–12) Similar findings have been observed in animal models, such as NC/Nga mice, a model of atopic dermatitis model,13,14) and in dry skin model mice.15,16) Such increases in nerve density are also found in the dermis of patients with atopic dermatitis and psoriasis.17,18) These findings are indicative of increases in sensory nerve fibers responsive to exogenous triggering factors and to various endogenous pruritogens from cutaneous cells, such as immune cells and keratinocytes, suggesting that hyperinnervation is partly responsible for itch sensitization.
3. MECHANISMS REGULATING SENSORY NERVE DENSITY IN THE SKIN
Epidermal hyperinnervation is probably caused by an imbalance in nerve elongation factors, such as nerve growth factor (NGF), and nerve repulsion factors, such as semaphorin 3A (Sema3A), produced by keratinocytes7) (Figs. 1C, D). These axonal guidance molecules may also act on keratinocytes, immune cells and vascular endothelial cells, and may be indirectly involved in the modulation of itching.
3.1. Nerve Elongation FactorsNerve Growth Factor (NGF)Keratinocyte-derived NGF is a major mediator of cutaneous innervation density, in that local NGF concentrations are higher in the lesional skin of patients with atopic dermatitis, psoriasis, prurigo nodularis, contact dermatitis and xerosis than in normal skin.5) In adult rat primary sensory neurons, NGF has been shown to upregulate neuropeptides, especially substance P and calcitonin-gene-related peptide (CGRP),19) both of which are involved in the hypersensitivity of itch sensation and neurogenic inflammation.20) Using a co-culture model of porcine DRG neurons and human skin cells, human atopic keratinocytes were found to produce elevated levels of NGF and to mediate an increased outgrowth of CGRP-immunoreactive fibers, whereas human atopic fibroblasts did not mediate this outgrowth.21) These findings indicate that keratinocytes are key factors in hyperinnervation in individuals with atopic dermatitis. Intradermal injection of NGF was also shown to sensitize nociceptors for cowhage- but not histamine-induced itch in human skin.22) Thus, increased NGF in the skin may sensitize primary afferents, thereby contributing to chronic itch such as atopic dermatitis. Interestingly, tumor necrosis factor (TNF)-α has been found to enhance NGF production in human keratinocytes.23) TNF-α is a pivotal proinflammatory cytokine in the innate immune response and a key molecule in skin inflammation. Mast cells have been identified as important potential sources of TNF-α.20) In addition, skin barrier disruption up-regulates TNF-α in the epidermis of acetone-treated mice, an acute dry skin model.24) These findings suggest that TNF-α is a positive regulator of NGF in the skin.
AmphiregulinAmphiregulin (AR), a protein belonging to the epidermal growth factor family, has been found to affect nerve outgrowth.25,26) AR expression was previously shown to be up-regulated in the epidermis of NC/Nga mice with atopic dermatitis-like symptoms,13) suggesting that AR may be involved in the modulation of epidermal nerve density. In addition, AR down-regulates expression of epithelial cell–cell junctional molecules such as E-cadherin and ZO-1 to construct adherens junctions and tight junctions, both of which are critical for skin barrier function.27) Indeed, the levels of expression of E-cadherin and ZO-1 were decreased in the epidermis of atopic NC/Nga mice, together with the increased expression of AR.13) Since AR affects the integrity of cell–cell junctions, these findings suggest the attenuation or abrogation of protective function against external mechanical, chemical and biological stimuli in inflammatory skin diseases.
ArteminArtemin is a glial cell line-derived neurotrophic factor. Artemin-expressing fibroblasts have been shown to accumulate in skin lesions of patients with atopic dermatitis.28) Moreover, dermal fibroblasts secrete artemin in response to substance P, released by cutaneous nerve fibers, inducing itch and/or neurogenic inflammation via the activation of neurokinin-1 (NK-1) receptor.29) Intradermal injection of artemin into mice resulted in peripheral nerve sprouting and thermal hyperalgesia.28) Therefore, artemin may be partly involved in hypersensitivity to warm sensations, mimicking warmth-provoked itch in atopic dermatitis.
3.2. Nerve Repulsion FactorsSemaphorin 3A (Sema3A)Sema3A is the first member of the semaphorin family shown to cause growth cone collapse in neurons, i.e., to function as an axonal repulsion factor, through its interaction with a neuropilin-1⁄plexin-A receptor complex.30) In addition, Sema3A inhibits NGF-induced sprouting of sensory afferents in adult rat spinal cord,31) whereas elevated levels of NGF reduce the Sema3A-induced collapse of sensory growth cones.32)
Several recent studies showed that Sema3A transcripts were expressed in cultured normal human epidermal keratinocytes.33,34) Sema3A proteins are mainly distributed in the suprabasal layer of normal human skin,33) consistent with findings showing that Sema3A is expressed in differentiated keratinocyte cultures.34) Moreover, epidermal Sema3A levels were lower in patients with atopic dermatitis than in healthy controls, while an increase in epidermal nerve density was found in the skin.33) Increased epidermal nerve density in acute dry skin mice was also associated with decreased levels of Sema3A expression,35,36) suggesting that decreasing the expression of Sema3A accelerates epidermal nerve growth in patients with dry skin condition such as atopic dermatitis and xerosis. Thus, epidermal innervation may be regulated by a fine balance between NGF and Sema3A.
Anosmin-1Anosmin-1 is an extracellular matrix glycoprotein encoded by the KAL1 (Kallmann syndrome 1 sequence) gene, the gene responsible for the X chromosome-linked recessive form of Kallmann syndrome.37) Anosmin-1 has been found to inhibit neurite outgrowth in cultured rat DRG neurons.38) KAL1 transcripts were also expressed in cultured keratinocytes and in normal human skin. Anosmin-1 was strongly expressed in the basal cell layer of normal skin, but its level of expression was lower in atopic skin, concomitant with an increase in epidermal nerve density. Thus, keratinocyte-derived anosmin-1 may be at least partly involved in modulating epidermal nerve density in patients with atopic dermatitis.38)
3.3. Roles of Matrix Metalloproteinase (MMP)-2 and MMP-8 in Cutaneous Nerve GrowthThe process of cutaneous nerve growth in pruritic skin such as atopic dermatitis requires several MMPs for growth cones to penetrate the three-dimensional extracellular matrix (ECM) barriers. Using in vitro models of ECM, such as Matrigel and type I collagen gel, MMP-2 localized on the growth cone was found to be involved in penetration into the basement membrane39) (Figs. 2A, B). In addition, MMP-8 secreted by nerve fibers was shown to be involved in nerve growth within the dermis, consisting mostly of types I and III collagens40) (Figs. 2A, C). The levels of expression of MMP-2 and MMP-8 were up-regulated by NGF and down-regulated by Sema3A, and both were induced by their enzymatic substrates, but not altered by non-substrate molecules. The selection and up-regulation of MMPs corresponding to the ECM components surrounding the growing nerve fibers may be required for efficient nerve fiber penetration, suggesting that the coordinated activation of neurotrophin and ECM-integrin signaling is necessary for efficient and long-distance axon extension.41,42) Since class 3 semaphorin signaling inhibits integrin-mediated adhesion signaling, Sema3A stimulation of growing nerve fibers may provide a reverse signaling pathway for these events.43)

4. ITCH-MEDIATING FIBERS IN THE PERIPHERY
4.1. Gastrin-Releasing Peptide (GRP)Since specific markers of itch-mediating fibers have not been identified to date, they could not be histologically identified in the periphery. Recent studies, however, have demonstrated that GRP receptor expressing cells mediate the itch sensation in the spinal cord.44) In addition, increases in cutaneous nerve fibers containing GRP have been observed in NC/Nga mice with atopic dermatitis-like symptoms.14) Therefore, GRP secreted from the central terminals of primary afferents may be involved in the transmission of itch signals in the spinal dorsal horn. Intradermal injections of GRP were found to elicit scratching behavior in mice, and the itch-related response was at least partly induced by the release of pruritogens through activation of bombesin receptors in mast cells.45) A more recent study showed that serum GRP levels correlate with pruritus in patients with atopic dermatitis.46) Thus, serum GRP level may be useful as a biomarker for itch and disease severity in patients with atopic dermatitis.
4.2. Mas-Related G-Protein Coupled Receptors (Mrgprs)Recent studies have demonstrated that the histamine-independent itch pathway involves members of the family of over 50 Mrgprs, especially MrgprAs, MrgprB4-5, MrgprC11 and MrgprD, which are restricted to small diameter DRG neurons in mice.47) Chloroquine and bovine adrenal medulla peptide 8-22 (BAM8-22) elicit itch-related scratching through MrgprA3 and MrgprC11, respectively, in mice.48) Both chloroquine and BAM8-22 also elicited itch in humans.49,50)
A more recent study using conditional transgenic mice revealed that ablation of MrgprA3+ DRG neurons led to substantial reductions in scratching evoked by multiple pruritogens and occurring spontaneously under chronic itch conditions.51) However, pain sensitivity remained intact in these mice. Moreover, mice in which transient receptor potential vanilloid 1 (TRPV1) was exclusively expressed in MrgprA3+ DRG neurons exhibited itch, but not pain, behavior in response to capsaicin. Although MrgprA3+ DRG neurons were sensitive to noxious heat, activation of TRPV1 in these neurons by noxious heat did not alter pain behavior. These findings suggest that MrgprA3 defines a specific subpopulation of DRG neurons that mediate itch. In mouse skin, MrgprA3+ fibers exclusively innervated the epidermis and responded to multiple pruritogens, suggesting that they are peripheral itch-specific fibers.
5. ANTI-PRURITIC THERAPIES
5.1. Anti-NGF Antibody and NGF Receptor AntagonistsAnti-NGF approaches have been tried to treat itch of atopic dermatitis in NC/Nga mice. Intraperitoneal administration of anti-NGF neutralizing antibody to atopic NC/Nga mice significantly attenuated both the increased number of nerve fibers in the epidermis and scratching behavior, but did not ameliorate scratching that had already developed.52) Similarly, application of the TrkA antagonists AG879 and K252a to the nape of atopic NC/Nga mice significantly improved established dermatitis and scratching behavior and reduced the numbers of nerve fibers in the epidermis, suggesting the importance of NGF in the pathogenesis of atopic dermatitis-like skin lesions.53) Thus, NGF and its receptors may be among the antipruritic targets in pruritic skin diseases such as atopic dermatitis.
5.2. Sema3A Replacement TherapyRecombinant Sema3A replacement approaches (intradermal injection or ointment application) were found to significantly inhibit scratching behavior and to improve dermatitis in NC/Nga mice with atopic dermatitis-like symptoms compared with controls.54,55) The therapeutic efficacy of exogenous Sema3A on atopic dermatitis-like symptoms was greater than that of current agents, such as betamethasone and tacrolimus.55) Moreover, histological analyses showed decreases in (i) the numbers of epidermal nerve fibers; (ii) the numbers of inflammatory infiltrates; (iii) the production of cytokines; (iv) the density of dermal blood vessels; and (v) epidermal thickness in Sema3A-treated lesional skin.54,55) These findings suggest that exogenous Sema3A not only affects sensory nerve fibers, but other cells that express neuropilin-1, include immune system cells, endothelial cells and keratinocytes.56) Thus, Sema3A and its receptors are promising therapeutic targets for atopic dermatitis.
5.3. Effectiveness of Conventional Therapies on Axonal Guidance MoleculesSeveral existing therapies normalize abnormal levels of axonal guidance molecules such as NGF and Sema3A in pruritic skin, reducting epidermal nerve density.
5.4. Olopatadine (A Histamine H1-Receptor Antagonist)Oral administration of olopatadine hydrochloride, a histamine H1-receptor (H1R) antagonist, significantly suppressed scratching behavior, improved dermatitis, and inhibited neurite outgrowth in the lesional skin of mice with atopic dermatitis-like symptoms. Notably, olopatadine treatment increased Sema3A expression in the epidermis.57,58) Although it is unclear whether these effects are caused by specific blocking of H1R signaling, olopatadine may in part improve imbalances between NGF and Sema3A in the epidermis.
5.5. Neurotropin (NTP)NTP, a non-protein extract isolated from the inflamed skin of rabbits inoculated with vaccinia virus, is widely used in Japan and China to treat various chronic pain conditions.59) In clinical studies in Japan, NTP has been shown to have anti-pruritic effects in patients with eczema, dermatitis and urticaria,60) and in those undergoing hemodialysis.61) NTP was found to inhibit NGF-induced neurite outgrowth of rat DRG neurons in vitro62) and to significantly reduce intraepidermal nerve growth in acetone-treated mice, a model for acute dry skin.63) In the latter, NTP significantly up-regulated epidermal Sema3A mRNA, but had no effect on expression of epidermal NGF mRNA.63) Thus, although its mechanisms are as yet unknown, NTP may reduce epidermal nerve density by inducing expression of Sema3A in the epidermis, resulting in suppression of itch.
5.6. EmollientA recent study using dry skin mice showed that application of heparinoid cream resulted in greater improvements in epidermal nerve density and epidermal NGF levels than application of petrolatum, although heparinoid cream had no effect on epidermal Sema3A levels.35) In addition, the increase of epidermal nerve fibers was more reduced by the immediate than the delayed application of emollients to dry skin, suggesting that the prompt application of emollients is more effective in normalizing epidermal hyperinnervation and expression of axon guidance molecules.
5.7. UV-Based TherapyVarious types of UV-based therapy, including psoralen-UVA (PUVA) and narrow-band UVB, have been widely used to treat patients with skin inflammation such as atopic dermatitis and psoriasis.64) The UV-based therapies were shown to reduce the number of cutaneous nerve fibers, especially in the epidermis, in patients with atopic dermatitis and psoriasis, and to inhibit pruritus.65,66) Similar effects of UV-based therapy on epidermal nerve fibers were observed in dry skin mice.36) The imbalance between Sema3A and NGF levels in the epidermis was normalized by PUVA or narrow-band UVB treatment.36,66) A recent study showed that excimer lamp treatment was the most effective form of UV-based therapy for intraepidermal nerve fibers.36)
5.8. New Anti-pruritic DrugsNalfurafine Hydrochloride (A Selective Kappa-Opioid Receptor Agonist)Several studies have demonstrated that mu- and kappa-opioid systems play pivotal roles in modulation of itch at the central nervous system. It is generally thought that the mu-opioid system induces itch, whereas the kappa-opioid system suppresses itch at the central level.7,67) Recently, the effectiveness of nalfurafine hydrochloride (REMITCH®), a selective kappa-opioid receptor agonist, on hemodialysis-related uremic pruritus was validated in a Phase III, randomized double-blind placebo-controlled trial.68) This drug is also expected to make further contributions to the treatment of patients with intractable itch, such as those with cholestasis and atopic dermatitis.
Peripheral mu- and kappa-opioid systems may also play important roles in pruritus of atopic dermatitis.69–71) One study revealed that topical application of mu-opioid receptor antagonist (naltrexone) cream to the skin inhibited itch in patients with atopic dermatitis.70) A peripherally restricted kappa-opioid agonist, ICI 204,448, also antagonized chloroquine-evoked scratching in mice.69) Moreover, the kappa-opioid system was found to be down-regulated in the epidermis of patients with atopic dermatitis and psoriasis, whereas the mu-opioid system was at normal levels.11,71) Down-regulation of the mu-opioid system and restoration of the kappa-opioid system by PUVA treatment have been observed in patients with atopic dermatitis, concomitant with a reduction in itch.71) Thus, these findings suggest that mu-opioid receptor antagonists or kappa-opioid receptor agonists have antipruritic effects at the peripheral level.
5.9. Aprepitant (A Neurokinin-1 (NK-1) Receptor Antagonist)Substance P is a key mediator in pruritus.72) In the periphery, substance P released from cutaneous nerve fibers induces itch and/or neurogenic inflammation.29) In addition, a recent study supports a role for NK-1 receptor-expressing spinal neurons in itch.73) Current evidence suggests that substance P is partially involved in the spinal transmission of itch signals.74)
Aprepitant is an oral NK-1 receptor antagonist75) widely used as an antiemetic agent in patients with chemotherapy-induced nausea and vomiting.76) Aprepitant was also found effective against intractable pruritus associated with Sézary syndrome, a leukemic, cutaneous, epidermotropic T-cell lymphoma.77) In a recent clinical trial, 16 of 20 patients with chronic itch treated with aprepitant showed a significant reduction in itch.78) Thus, although some patients did not respond, these findings indicated that aprepitant is effective in patients with chronic itch, including atopic dermatitis.
5.10. Histamine H4 Receptor (H4R) AntagonistsH4R have been reported involved in histamine-evoked itch in animal models.79) While specific H4R agonists induced itch, pretreatment with the H4R antagonist JNJ7777120 inhibited this response, and histamine or H4R agonist-evoked itch was attenuated in H4R-deficient mice. Moreover, inhibiting both H1R and H4R almost completely eliminated histamine-evoked scratching.80) A more recent study showed that the H1R antagonist olopatadine and the H4R antagonist JNJ7777120 reduced scratching behavior and skin inflammation in the NC/Nga mouse model of chronic allergic dermatitis induced by repeated challenges with picryl chloride.58) Thus, H4R antagonists may be effective in patients with chronic itch, including those with atopic dermatitis.
6. CONCLUSION
This review presents recent knowledge regarding itch sensitization associated with epidermal nerve density controlled by nerve elongation factors (e.g., NGF) and nerve repulsion factors (e.g., Sema3A) through the regulation of expression of MMPs, especially in atopic dermatitis. In addition, treatment with anti-NGF agents, Sema3A replacement and other treatments such as UV-based therapies may normalize epidermal nerve fiber density. New substances and classes of antipruritic drugs are needed. The research on and development of anti-pruritic drugs may contribute to improvements in the quality of life of patients who suffer from intractable pruritus such as atopic dermatitis.
Acknowledgments
This work was supported by a Health Labor Sciences Research Grant for Research on Allergic Disease and Immunology from the Japanese Ministry of Health, Labour and Welfare, by a KAKENHI (20591354 and 2079081) and a “High-Tech Research Center” Project for Private Universities: matching fund subsidy from MEXT, and by a JSPS Research Fellow.
REFERENCES
- 1) Weisshaar E, Apfelbacher C, Jäger G, Zimmermann E, Bruckner T, Diepgen TL, Gollnick H. Pruritus as a leading symptom: clinical characteristics and quality of life in German and Ugandan patients. Br. J. Dermatol., 155, 957–964 (2006).
- 2) Pisoni RL, Wikström B, Elder SJ, Akizawa T, Asano Y, Keen ML, Saran R, Mendelssohn DC, Young EW, Port FK. Pruritus in haemodialysis patients: International results from the Dialysis Outcomes and Practice Patterns Study (DOPPS). Nephrol. Dial. Transplant., 21, 3495–3505 (2006).
- 3) Picardi A, Lega I, Tarolla E. Suicide risk in skin disorders. Clin. Dermatol., 31, 47–56 (2013).
- 4) Murota H, Kitaba S, Tani M, Wataya-Kaneda M, Azukizawa H, Tanemura A, Umegaki N, Terao M, Kotobuki Y, Katayama I. Impact of sedative and non-sedative antihistamines on the impaired productivity and quality of life in patients with pruritic skin diseases. Allergol. Int., 59, 345–354 (2010).
- 5) Ikoma A, Steinhoff M, Ständer S, Yosipovitch G, Schmelz M. The neurobiology of itch. Nat. Rev. Neurosci., 7, 535–547 (2006).
- 6) Basbaum AI, Bautista DM, Scherrer G, Julius D. Cellular and molecular mechanisms of pain. Cell, 139, 267–284 (2009).
- 7) Tominaga M, Takamori K. Recent advances in pathophysiological mechanisms of itch. Expert Rev. Dermatol., 5, 197–212 (2010).
- 8) Akiyama T, Tominaga M, Davoodi A, Nagamine M, Blansit K, Horwitz A, Carstens MI, Carstens E. Cross-sensitization of histamine-independent itch in mouse primary sensory neurons. Neuroscience, 226, 305–312 (2012).
- 9) Akiyama T, Carstens MI, Carstens E. Enhanced scratching evoked by PAR-2 agonist and 5-HT but not histamine in a mouse model of chronic dry skin itch. Pain, 151, 378–383 (2010).
- 10) Schuhknecht B, Marziniak M, Wissel A, Phan NQ, Pappai D, Dangelmaier J, Metze D, Ständer S. Reduced intraepidermal nerve fibre density in lesional and nonlesional prurigo nodularis skin as a potential sign of subclinical cutaneous neuropathy. Br. J. Dermatol., 165, 85–91 (2011).
- 11) Taneda K, Tominaga M, Negi O, Tengara S, Kamo A, Ogawa H, Takamori K. Evaluation of epidermal nerve density and opioid receptor levels in psoriatic itch. Br. J. Dermatol., 165, 277–284 (2011).
- 12) Kou K, Nakamura F, Aihara M, Chen H, Seto K, Komori-Yamaguchi J, Kambara T, Nagashima Y, Goshima Y, Ikezawa Z. Decreased expression of semaphorin-3A, a neurite-collapsing factor, is associated with itch in psoriatic skin. Acta Derm. Venereol., 92, 521–528 (2012).
- 13) Tominaga M, Ozawa S, Ogawa H, Takamori K. A hypothetical mechanism of intraepidermal neurite formation in NC/Nga mice with atopic dermatitis. J. Dermatol. Sci., 46, 199–210 (2007).
- 14) Tominaga M, Ogawa H, Takamori K. Histological characterization of cutaneous nerve fibers containing gastrin-releasing peptide in NC/Nga mice: an atopic dermatitis model. J. Invest. Dermatol., 129, 2901–2905 (2009).
- 15) Miyamoto T, Nojima H, Shinkado T, Nakahashi T, Kuraishi Y. Itch-associated response induced by experimental dry skin in mice. Jpn. J. Pharmacol., 88, 285–292 (2002).
- 16) Tominaga M, Ozawa S, Tengara S, Ogawa H, Takamori K. Intraepidermal nerve fibers increase in dry skin of acetone-treated mice. J. Dermatol. Sci., 48, 103–111 (2007).
- 17) Urashima R, Mihara M. Cutaneous nerves in atopic dermatitis. A histological, immunohistochemical and electron microscopic study. Virchows Arch., 432, 363–370 (1998).
- 18) Nakamura M, Toyoda M, Morohashi M. Pruritogenic mediators in psoriasis vulgaris: comparative evaluation of itch-associated cutaneous factors. Br. J. Dermatol., 149, 718–730 (2003).
- 19) Verge VM, Richardson PM, Wiesenfeld-Hallin Z, Hökfelt T. Differential influence of nerve growth factor on neuropeptide expression in vivo: a novel role in peptide suppression in adult sensory neurons. J. Neurosci., 15, 2081–2096 (1995).
- 20) Steinhoff M, Ständer S, Seeliger S, Ansel JC, Schmelz M, Luger T. Modern aspects of cutaneous neurogenic inflammation. Arch. Dermatol., 139, 1479–1488 (2003).
- 21) Roggenkamp D, Falkner S, Stäb F, Petersen M, Schmelz M, Neufang G. Atopic keratinocytes induce increased neurite outgrowth in a coculture model of porcine dorsal root ganglia neurons and human skin cells. J. Invest. Dermatol., 132, 1892–1900 (2012).
- 22) Rukwied RR, Main M, Weinkauf B, Schmelz M. NGF sensitizes nociceptors for cowhage- but not histamine-induced itch in human skin. J. Invest. Dermatol., 133, 268–270 (2013).
- 23) Takaoka K, Shirai Y, Saito N. Inflammatory cytokine tumor necrosis factor-alpha enhances nerve growth factor production in human keratinocytes, HaCaT cells. J. Pharmacol. Sci., 111, 381–391 (2009).
- 24) Wood LC, Jackson SM, Elias PM, Grunfeld C, Feingold KR. Cutaneous barrier perturbation stimulates cytokine production in the epidermis of mice. J. Clin. Invest., 90, 482–487 (1992).
- 25) Kimura H, Schubert D. Schwannoma-derived growth factor promotes the neuronal differentiation and survival of PC12 cells. J. Cell Biol., 116, 777–783 (1992).
- 26) Nilsson A, Kanje M. Amphiregulin acts as an autocrine survival factor for adult sensory neurons. Neuroreport, 16, 213–218 (2005).
- 27) Chung E, Cook PW, Parkos CA, Park YK, Pittelkow MR, Coffey RJ. Amphiregulin causes functional downregulation of adherens junctions in psoriasis. J. Invest. Dermatol., 124, 1134–1140 (2005).
- 28) Murota H, Izumi M, Abd El-Latif MI, Nishioka M, Terao M, Tani M, Matsui S, Sano S, Katayama I. Artemin causes hypersensitivity to warm sensation, mimicking warmth-provoked pruritus in atopic dermatitis. J. Allergy Clin. Immunol., 130, 671–682, e4 (2012).
- 29) Paus R, Schmelz M, Bíró T, Steinhoff M. Frontiers in pruritus research: scratching the brain for more effective itch therapy. J. Clin. Invest., 116, 1174–1186 (2006).
- 30) Fujisawa H. Discovery of semaphorin receptors, neuropilin and plexin, and their functions in neural development. J. Neurobiol., 59, 24–33 (2004).
- 31) Tang XQ, Tanelian DL, Smith GM. Semaphorin 3A inhibits nerve growth factor-induced sprouting of nociceptive afferents in adult rat spinal cord. J. Neurosci., 24, 819–827 (2004).
- 32) Dontchev VD, Letourneau PC. Nerve growth factor and semaphorin 3A signaling pathways interact in regulating sensory neuronal growth cone motility. J. Neurosci., 22, 6659–6669 (2002).
- 33) Tominaga M, Ogawa H, Takamori K. Decreased production of semaphorin 3A in the lesional skin of atopic dermatitis. Br. J. Dermatol., 158, 842–844 (2008).
- 34) Fukamachi S, Bito T, Shiraishi N, Kobayashi M, Kabashima K, Nakamura M, Tokura Y. Modulation of semaphorin 3A expression by calcium concentration and histamine in human keratinocytes and fibroblasts. J. Dermatol. Sci., 61, 118–123 (2011).
- 35) Kamo A, Tominaga M, Negi O, Tengara S, Ogawa H, Takamori K. Topical application of emollients prevents dry skin-inducible intraepidermal nerve growth in acetone-treated mice. J. Dermatol. Sci., 62, 64–66 (2011).
- 36) Kamo A, Tominaga M, Tengara S, Ogawa H, Takamori K. Inhibitory effects of UV-based therapy on dry skin-inducible nerve growth in acetone-treated mice. J. Dermatol. Sci., 62, 91–97 (2011).
- 37) Soussi-Yanicostas N, Hardelin JP, Arroyo-Jimenez MM, Ardouin O, Legouis R, Levilliers J, Traincard F, Betton JM, Cabanié L, Petit C. Initial characterization of anosmin-1, a putative extracellular matrix protein synthesized by definite neuronal cell populations in the central nervous system. J. Cell Sci., 109, 1749–1757 (1996).
- 38) Tengara S, Tominaga M, Kamo A, Taneda K, Negi O, Ogawa H, Takamori K. Keratinocyte-derived anosmin-1, an extracellular glycoprotein encoded by the X-linked Kallmann syndrome gene, is involved in modulation of epidermal nerve density in atopic dermatitis. J. Dermatol. Sci., 58, 64–71 (2010).
- 39) Tominaga M, Kamo A, Tengara S, Ogawa H, Takamori K. In vitro model for penetration of sensory nerve fibres on a Matrigel basement membrane: implications for possible application to intractable pruritus. Br. J. Dermatol., 161, 1028–1037 (2009).
- 40) Tominaga M, Tengara S, Kamo A, Ogawa H, Takamori K. Matrix metalloproteinase-8 is involved in dermal nerve growth: implications for possible application to pruritus from in vitro models. J. Invest. Dermatol., 131, 2105–2112 (2011).
- 41) Grabham PW, Goldberg DJ. Nerve growth factor stimulates the accumulation of beta1 integrin at the tips of filopodia in the growth cones of sympathetic neurons. J. Neurosci., 17, 5455–5465 (1997).
- 42) Gardiner NJ. Integrins and the extracellular matrix: key mediators of development and regeneration of the sensory nervous system. Dev. Neurobiol., 71, 1054–1072 (2011).
- 43) Zhou Y, Gunput RA, Pasterkamp RJ. Semaphorin signaling: progress made and promises ahead. Trends Biochem. Sci., 33, 161–170 (2008).
- 44) Sun YG, Chen ZF. A gastrin-releasing peptide receptor mediates the itch sensation in the spinal cord. Nature, 448, 700–703 (2007).
- 45) Andoh T, Kuwazono T, Lee JB, Kuraishi Y. Gastrin-releasing peptide induces itch-related responses through mast cell degranulation in mice. Peptides, 32, 2098–2103 (2011).
- 46) Kagami S, Sugaya M, Suga H, Morimura S, Kai H, Ohmatsu H, Fujita H, Tsunemi Y, Sato S. Serum gastrin-releasing peptide levels correlate with pruritus in patients with atopic dermatitis. J. Invest. Dermatol., 133, 1673–1675 (2013).
- 47) Dong X, Han S, Zylka MJ, Simon MI, Anderson DJ. A diverse family of GPCRs expressed in specific subsets of nociceptive sensory neurons. Cell, 106, 619–632 (2001).
- 48) Liu Q, Tang Z, Surdenikova L, Kim S, Patel KN, Kim A, Ru F, Guan Y, Weng HJ, Geng Y, Undem BJ, Kollarik M, Chen ZF, Anderson DJ, Dong X. Sensory neuron-specific GPCR Mrgprs are itch receptors mediating chloroquine-induced pruritus. Cell, 139, 1353–1365 (2009).
- 49) Abila B, Ezeamuzie IC, Igbigbi PS, Ambakederemo AW, Asomugha L. Effects of two antihistamines on chloroquine and histamine induced weal and flare in healthy African volunteers. Afr. J. Med. Med. Sci., 23, 139–142 (1994).
- 50) Sikand P, Dong X, LaMotte RH. BAM8-22 peptide produces itch and nociceptive sensations in humans independent of histamine release. J. Neurosci., 31, 7563–7567 (2011).
- 51) Han L, Ma C, Liu Q, Weng HJ, Cui Y, Tang Z, Kim Y, Nie H, Qu L, Patel KN, Li Z, McNeil B, He S, Guan Y, Xiao B, Lamotte RH, Dong X. A subpopulation of nociceptors specifically linked to itch. Nat. Neurosci., 16, 174–182 (2013).
- 52) Takano N, Sakurai T, Kurachi M. Effects of anti-nerve growth factor antibody on symptoms in the NC/Nga mouse, an atopic dermatitis model. J. Pharmacol. Sci., 99, 277–286 (2005).
- 53) Takano N, Sakurai T, Ohashi Y, Kurachi M. Effects of high-affinity nerve growth factor receptor inhibitors on symptoms in the NC/Nga mouse atopic dermatitis model. Br. J. Dermatol., 156, 241–246 (2007).
- 54) Yamaguchi J, Nakamura F, Aihara M, Yamashita N, Usui H, Hida T, Takei K, Nagashima Y, Ikezawa Z, Goshima Y. Semaphorin 3A alleviates skin lesions and scratching behavior in NC/Nga mice, an atopic dermatitis model. J. Invest. Dermatol., 128, 2842–2849 (2008).
- 55) Negi O, Tominaga M, Tengara S, Kamo A, Taneda K, Suga Y, Ogawa H, Takamori K. Topically applied semaphorin 3A ointment inhibits scratching behavior and improves skin inflammation in NC/Nga mice with atopic dermatitis. J. Dermatol. Sci., 66, 37–43 (2012).
- 56) Romeo PH, Lemarchandel V, Tordjman R. Neuropilin-1 in the immune system. Adv. Exp. Med. Biol., 515, 49–54 (2002).
- 57) Murota H, El-latif MA, Tamura T, Amano T, Katayama I. Olopatadine hydrochloride improves dermatitis score and inhibits scratch behavior in NC/Nga mice. Int. Arch. Allergy Immunol., 153, 121–132 (2010).
- 58) Ohsawa Y, Hirasawa N. The antagonism of histamine H1 and H4 receptors ameliorates chronic allergic dermatitis via anti-pruritic and anti-inflammatory effects in NC/Nga mice. Allergy, 67, 1014–1022 (2012).
- 59) Yoshii H, Suehiro S, Watanabe K, Yanagihara Y. Immunopharmacological actions of an extract isolated from inflamed skin of rabbits inoculated with vaccinia virus (neurotropin); enhancing effect on delayed type hypersensitivity response through the induction of Lyt-1+2-T cells. Int. J. Immunopharmacol., 9, 443–451 (1987).
- 60) Clinical Research Group for Neurotropin. Double blind study on antiprurigous effect of neurotropin on chronic uriticaria and eczematous dermatitis. Nishinihon J. Dermatol., 41, 552–559 (1979).
- 61) Yago H, Fujita Y, Kaku H, Naka F, Nishikawa K, Kawakubo H, Nakano K, Matsumoto K, Suehiro S. Study on pruritus in hemodialysis patients and the antipruritic effect of neurotropin: plasma levels of C3a, C5a, bradykinin and lipid peroxides. Nippon Jinzo Gakkai Shi, 31, 1061–1067 (1989).
- 62) Taneda K, Tominaga M, Tengara S, Ogawa H, Takamori K. Neurotropin inhibits both capsaicin-induced substance P release and nerve growth factor-induced neurite outgrowth in cultured rat dorsal root ganglion neurones. Clin. Exp. Dermatol., 35, 73–77 (2010).
- 63) Kamo A, Tominaga M, Taneda K, Ogawa H, Takamori K. Neurotropin inhibits the increase in intraepidermal nerve density in the acetone-treated dry-skin mouse model. doi: (2013).
- 64) Krutmann J. Phototherapy for atopic dermatitis. Clin. Exp. Dermatol., 25, 552–558 (2000).
- 65) Wallengren J, Sundler F. Phototherapy reduces the number of epidermal and CGRP-positive dermal nerve fibres. Acta Derm. Venereol., 84, 111–115 (2004).
- 66) Tominaga M, Tengara S, Kamo A, Ogawa H, Takamori K. Psoralen-ultraviolet A therapy alters epidermal Sema3A and NGF levels and modulates epidermal innervation in atopic dermatitis. J. Dermatol. Sci., 55, 40–46 (2009).
- 67) Phan NQ, Lotts T, Antal A, Bernhard JD, Ständer S. Systemic kappa opioid receptor agonists in the treatment of chronic pruritus: a literature review. Acta Derm. Venereol., 92, 555–560 (2012).
- 68) Kumagai H, Ebata T, Takamori K, Miyasato K, Muramatsu T, Nakamoto H, Kurihara M, Yanagita T, Suzuki H. Efficacy and safety of a novel κ-agonist for managing intractable pruritus in dialysis patients. Am. J. Nephrol., 36, 175–183 (2012).
- 69) Inan S, Cowan A. Kappa opioid agonists suppress chloroquine-induced scratching in mice. Eur. J. Pharmacol., 502, 233–237 (2004).
- 70) Bigliardi PL, Stammer H, Jost G, Rufli T, Büchner S, Bigliardi-Qi M. Treatment of pruritus with topically applied opiate receptor antagonist. J. Am. Acad. Dermatol., 56, 979–988 (2007).
- 71) Tominaga M, Ogawa H, Takamori K. Possible roles of epidermal opioid systems in pruritus of atopic dermatitis. J. Invest. Dermatol., 127, 2228–2235 (2007).
- 72) Wallengren J. Neuroanatomy and neurophysiology of itch. Dermatol. Ther., 18, 292–303 (2005).
- 73) Carstens EE, Carstens MI, Simons CT, Jinks SL. Dorsal horn neurons expressing NK-1 receptors mediate scratching in rats. Neuroreport, 21, 303–308 (2010).
- 74) Akiyama T, Tominaga M, Davoodi A, Nagamine M, Blansit K, Horwitz A, Carstens MI, Carstens E. Roles for substance P and gastrin-releasing peptide as neurotransmitters released by primary afferent pruriceptors. J. Neurophysiol., 109, 742–748 (2013).
- 75) Quartara L, Altamura M. Tachykinin receptors antagonists: from research to clinic. Curr. Drug Targets, 7, 975–992 (2006).
- 76) Hesketh PJ. Chemotherapy-induced nausea and vomiting. N. Engl. J. Med., 358, 2482–2494 (2008).
- 77) Duval A, Dubertret L. Aprepitant as an antipruritic agent? N. Engl. J. Med., 361, 1415–1416 (2009).
- 78) Ständer S, Siepmann D, Herrgott I, Sunderkötter C, Luger TA. Targeting the neurokinin receptor 1 with aprepitant: a novel antipruritic strategy. PLoS ONE, 5, e10968 (2010).
- 79) Thurmond RL, Gelfand EW, Dunford PJ. The role of histamine H1 and H4 receptors in allergic inflammation: the search for new antihistamines. Nat. Rev. Drug Discov., 7, 41–53 (2008).
- 80) Dunford PJ, Williams KN, Desai PJ, Karlsson L, McQueen D, Thurmond RL. Histamine H4 receptor antagonists are superior to traditional antihistamines in the attenuation of experimental pruritus. J. Allergy Clin. Immunol., 119, 176–183 (2007).