RESULTS AND DISCUSSION
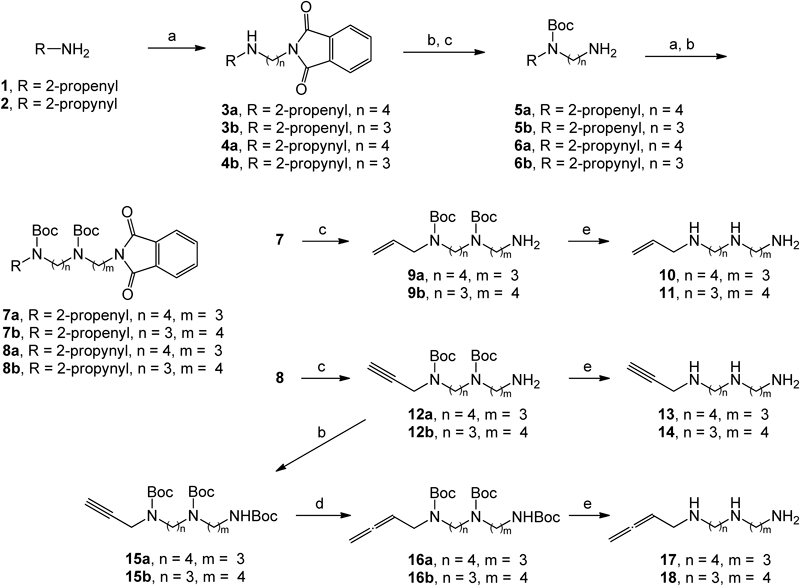
Synthesis of N8- or N1-Substituted Spermidine DerivativesWe tried a modified synthetic route that had been reported by Bey et al.,2) but we found it unsuitable for the synthesis of spermidine derivatives. An alternative synthetic route to introduce the propenyl, propynyl, or butadienyl functional groups on the terminal amine (either N1 or N8) of the unsymmetrical polyamine spermidine is shown in Chart 1. We used KF-Celite in the synthesis,12) which has been successfully used previously for polyamine synthesis.13) Acetonitrile solutions of 2-propenylamine 1 or 2-propynylamine 2 and N-(4-bromobutyl)phthalimide (for N8-substitution) or N-(3-bromopropyl)phthalimide (for N1-substitution) were refluxed with KF-Celite to obtain compound 3 or 4. Compounds 5 and 6, which were prepared via tert-butoxycarbonyl (Boc) protection under the usual conditions, followed by hydrazine degradation, were similarly refluxed in acetonitrile with nearly equivalent amount of N-(3-bromopropyl)phthalimide or N-(4-bromobutyl)phthalimide with KF-Celite to obtain compound 7 or 8 containing a spermidine skeleton. Hydrochlorides of N-(3-aminopropyl)-N′-2-propenyl-1,4-butanediamine (N8-propenyl spermidine, designated below as N8-propenyl Spd, 10), N-[3-(2-propenylamino)propyl]-1,4-butanediamine (N1-propenyl spermidine, designated below as N1-propenyl Spd, 11), N-(3-aminopropyl)-N′-2-propynyl-1,4-butanediamine (N8-propynyl spermidine, designated below as N8-propynyl Spd, 13), and N-[3-(2-propynylamino)propyl]-1,4-butanediamine (N1-propynyl spermidine, designated below as N1-propynyl Spd, 14) precipitated in reasonable yield following the standard work-up for the sequential removal of the phthaloyl and Boc groups of compounds 7 and 8. The hydrochlorides of N-(3-aminopropyl)-N′-2,3-butadienyl-1,4-butanediamine (N8-butadienyl spermidine, designated below as N8-butadienyl Spd, 17) and N-[3-(2,3-butadienylamino)propyl]-1,4-butanediamine (N1-butadienyl spermidine, designated below as N1-butadienyl Spd, 18) precipitated similarly after conversion of the 2-propynyl group of tri-Boc-protected compound 15 to a butadienyl group according to the method reported by Casara et al.14) Of the six spermidine derivatives synthesized, N8-butadienyl Spd alone has been listed in a patent specification by Bey.15) However, the preparation and characterization of this compound have not been reported in the literature.
Inactivation of SMO and APAOEnzyme activities were measured by mass spectrometry as previously reported,5) [5,8-13C2,4,8-15N2]spermidine (13C2,15N2-spermidine) was measured by using [5,8-13C2,4,9-15N2]spermine (13C2,15N2-spermine) and [5,8-13C2,4,9-15N2]-N1-acetylspermine (13C2,15N2-N1-acetylspermine) as substrates for SMO and APAO, respectively. The apparent Km value of purified SMO was 120 µM (pH 9.0), consistent with the reported value of 190 µM (pH 8.3).16) The apparent Km value of purified APAO was 1.7 µM (pH 9.0), consistent with the reported value of 0.85 µM (pH 8.0).17)
To examine whether the synthetic compounds, N1- and N8-butadienyl Spd, N1- and N8-propenyl Spd, and N1- and N8-propynyl Spd inhibit SMO and APAO activity, the enzymes were pre-incubated at pH 9.0 for 15 min with 1 µM synthetic compound, and the remaining activity was measured under standard assay conditions (Fig. 1). SMO activity was potently inhibited by N8-butadienyl Spd and moderately inhibited by N1-butadienyl Spd. MDL 72527 and the other four compounds only marginally inhibited SMO activity or failed to inhibit SMO at this concentration. In contrast, APAO activity was strongly inhibited by MDL 72527, N1-butadienyl Spd and N8-butadienyl Spd, but was only moderately inhibited by N1- and N8-propenyl Spd. N1- and N8-propynyl Spd exhibited no inhibitory activity toward APAO. Pre-incubation at pH 7.0 and 8.0 gave very similar results with respect to the relative potencies of these inhibitors and their selectivities toward SMO and APAO. These results suggest that the SMO active site discriminates between these various compounds more strictly than the APAO active site. This is consistent with previous observations reported in the literature.3,11,18) Based on these results, N1- and N8-butadienyl Spd were selected for further study.

Time courses of the SMO and APAO reactions in the presence of N1- or N8-butadienyl Spd were monitored without pre-incubating the enzymes with the inhibitors. In the absence of the inhibitors, the reaction proceeded linearly with time. The rate of production of 13C2,15N2-spermidine from 13C2,15N2-spermine by SMO and from 13C2,15N2-N1-acetylspermine by APAO gradually decreased in the presence of the inhibitors over a period of 5 to 30 min, depending on the concentrations of the inhibitors (Fig. 2). These results indicated the gradual loss of enzyme activity. Dialysis of the incubation mixture 30 min after the beginning of the reaction in the presence of 10 µM N8-butadienyl Spd did not restore either the SMO activity or the APAO activity (data not shown), which confirms that the enzymes were irreversibly inactivated. N8-Butadienyl Spd more potently inactivated SMO than N1-butadienyl Spd at the same concentration, whereas N1-butadienyl Spd was slightly more effective than N8-butadienyl Spd at inactivating APAO.

We next examined whether spermidine was liberated from N1- and N8-butadienyl Spd during incubation with different amounts of APAO and SMO. Figure 3 shows that spermidine was produced from either N1- or N8-butadienyl Spd in an amount roughly proportional to the amount of APAO used. The rate of spermidine production rapidly decreased with time, indicating that the enzyme became progressively more inactivated. The apparent molar proportions of spermidine to APAO were approximately 60 and 80 for N1- and N8-butadienyl Spd, respectively, which were calculated by extrapolating to the point of complete inactivation. These results are consistent with the notion that the two inhibitors are suicide substrates of APAO.19) In contrast, little (if any) spermidine was liberated upon incubation of SMO with either N1- or N8-butadienyl Spd, even though the enzyme was irreversibly inhibited during the incubation (data not shown).

Although the precise mechanism of SMO inactivation by N8-butadienyl Spd remains to be elucidated, the molecular structure of N8-butadienyl Spd strongly suggests that it fits well into the active site of the enzyme. Covalent binding to the flavin moiety in the active site of APAO was proposed as part of the inactivation mechanism of MDL 72527.20) Crystallization and X-ray crystallography will facilitate the elucidation of the precise mechanism of inactivation of SMO by N8-butadienyl Spd.
The inhibitory effects of N1- and N8-butadienyl Spd and MDL 72527 on SMO and APAO were compared with or without preincubation, and the results are summarized in Table 1 as the concentrations producing 50% inhibition. N8-Butadienyl Spd was approximately 50-fold more effective than MDL 72527 at inhibiting SMO. The inhibition of SMO with the three inhibitors decreased by approximately 10-fold in the presence of 0.2 mM spermine (without preincubation), whereas the inhibition of APAO was decreased to a much greater extent, approximately 700-fold with MDL 72527, 150-fold with N1-butadienyl Spd, and 90-fold with N8-butadienyl Spd in the presence of 0.2 mM N1-acetylspermine (without preincubation). Thus, N1- and N8-butadienyl Spd inactivated APAO two- to three-fold more effectively than MDL 72527 under the test conditions. This is most likely because the substrates of SMO and APAO compete with the inhibitors for access to the substrate binding site. In fact, 1 µM N8-butadienyl Spd almost completely inactivated SMO in 15 min in the absence of substrate (Fig. 1), whereas in the presence of 0.2 mM substrate, approximately 50% of the initial activity still remained after incubation for 15 min (Fig. 2).
Table 1. Concentrations of MDL 72527, and
N1- or
N8-Butadienyl Spd Resulting in 50% Inhibition of SMO and APAO Activity
| SMO | APAO |
|---|
| Inhibitor | Preincubation |
|---|
| Yes | No | Yes | No |
|---|
| Concentration resulting in 50% inhibition (μM) |
|---|
| MDL72527 | 6.1 | 57.4 | 0.02 | 13.1 |
| N1-Butadienyl Spd | 0.8 | 8.3 | 0.03 | 4.7 |
| N8-Butadienyl Spd | 0.1 | 1.8 | 0.05 | 4.3 |
Foot note: Enzyme activity was determined at various inhibitor concentrations either with (“Yes”) or without (“No”) preincubation of the enzymes with inhibitors (mean of duplicate determinations for each concentration are shown). The activity vs. inhibitor concentration curve was used to estimate the concentration (µM) resulting in 50% inhibition. For preincubation, the enzyme was incubated with the inhibitor for 15 min at 37°C in the absence of substrate. An appropriate substrate was then added, and the incubation was continued for an additional 30 min under standard assay conditions as described in Materials and Methods.
These results suggest that N8-butadienyl Spd could be used to study the biological significance of SMO and APAO, although it is difficult to inhibit SMO and APAO separately. It might be possible to discriminate between SMO and APAO activity, because SMO might be inactivated more easily than APAO at a low concentration of N8-butadienyl Spd in biological samples, that contain the native substrates for these enzymes. The detection of 15N2-labeled spermidine, liberated from 15N2-labeled N8-butadienyl Spd, by mass spectrometry may serve as an indicator of APAO inactivation. The labeled compound could be synthesized according to the procedures described in this report, by using 15N-labeled N-(3-bromopropyl)- and N-(4-bromobutyl)phthalimide as starting materials.
MATERIALS AND METHODS
General MethodsProcedure A (amine alkylation): The amine (1–2.5 mmol) and N-3-(bromopropyl)- or N-4-(bromobutyl)phthalimide (1 mmol) were dissolved in acetonitrile (5 mL) and refluxed with KF-Celite (0.3 g) with constant stirring for 3–16 h. KF-Celite was thoroughly dried over P2O5 beforehand. After evaporation of the filtrate, the residue was purified through a silica gel column. Procedure B (Boc protection): The amine (1 mmol), triethylamine (0.3 mL), and (Boc)2O (0.23 mL, 1.06 mmol) were dissolved in acetonitrile (6 mL) and stirred at room temperature for 12 h. The solvent was evaporated in vacuo, and the residue was dissolved in Et2O and washed successively with 1 M KHSO4, 1 M NaHCO3, and brine. The organic phase was evaporated after drying over MgSO4. The obtained residue was used for the next reaction. Procedure C (hydrazine degradation): The compound (1 mmol) and hydrazine monohydrate (0.3 mL) were dissolved in MeOH (7 mL) and refluxed for 3 h. The solvent was evaporated in vacuo, and the residue was extracted with 4 M ammonia and chloroform. The organic phase was evaporated in vacuo. The obtained residue was used for the next reaction. Procedure D (removal of Boc): The tri-Boc spermidine derivative (1 mmol) was dissolved in EtOH (12 mL) and 0.5 M HCl (ethanolic) (12 mL) and stirred at 50°C for 12 h. The hydrochloride of the spermidine derivative precipitated as white crystals, and was recrystallized from aqueous EtOH.
Experimental details are presented below for compounds 17 and 18 in Chart 1, which represent compounds 10, 11, 13, and 14. Compounds 3, 5, 7, and 9 were prepared using a similar method. The apparent yields of the oily intermediates were calculated by weight and confirmed by the observation of a major single band in TLC. The final six hydrochlorides of the spermidine derivatives showed purities above 95%. All solvents and chemicals were used as purchased unless otherwise stated.
2-[4-(2-Propynylamino)butyl]-1H-isoindole-1,3(2H)-dione (4a)2-Propynylamine 2 (0.65 mL, 10 mmol) was treated with N-(4-bromobutyl)phthalimide (1.13 g, 4 mmol) for 3 h according to Procedure A, and purification on a silica gel (8 g) column eluting with benzene–acetone (10 : 1), afforded compound 4a (0.75 g, 74%).
2-[3-(2-Propynylamino)propyl]-1H-isoindole-1,3(2H)-dione (4b)With the exception of the use of N-(3-bromopropyl)phthalimide (1.07 g, 4 mmol), all treatments were the same as those described for compound 4a and b (0.75 g, 78%).
2-[3-[(tert-Butoxycarbonyl)[4-[(tert-butoxycarbonyl)-(2-propynyl)amino]butyl]amino]propyl]-1H-isoindole-1,3(2H)-dione (8a)Compound 4a (0.75 g, 2.9 mmol) was treated according to Procedure B, and the resulting Boc-protected 4a (1.0 g, 2.86 mmol) was treated according to Procedure C to obtain compound 6a (0.6 g, 2.65 mmol). This compound was treated with N-(3-bromopropyl)phthalimide (0.75 g, 2.8 mmol) for 3 h according to Procedure A, and purification using a silica gel (9 g) column eluting with CHCl3–MeOH–AcOH (20 : 1 : 0.1) afforded the pure product. This product (0.66 g, 1.6 mmol) was treated according to Procedure B to obtain compound 8a (0.76 g, 52% yield for 3 steps).
2-[4-[(tert-Butoxycarbonyl)[3-[(tert-butoxycarbonyl)-(2-propynyl)amino]propyl]amino]butyl]-1H-isoindole-1,3(2H)-dione (8b)Compound 4b (0.75 g, 3.1 mmol) was treated according to Procedure B, and the Boc-protected 4b (1.0 g, 3.0 mmol) was treated according to Procedure C to obtain compound 6b (0.59 g, 2.8 mmol). Compound 6b was similarly reacted with N-(4-bromobutyl)phthalimide (0.85 g, 3.0 mmol) and purified by the same chromatography scheme used to obtain compound 8a. The purified product (0.76 g, 1.6 mmol) was treated according to Procedure B to obtain compound 8b (0.68 g, 43% yield for 3 steps).
N-2,3-Butadienyl-N,N′-bis(tert-butoxycarbonyl)-N′-[3-[(tert-butoxycarbonyl)amino]propyl]-1,4-butanediamine (16a)Compound 8a (0.76 g, 1.5 mmol) was treated according to Procedure C to obtain compound 12a (0.49 g, 1.3 mmol). Subsequently, compound 12a was treated using Procedure B to obtain the tri-Boc-protected derivative 15a (0.62 g, 1.27 mmol), which was refluxed with 37% formalin (0.2 mL), diisopropylamine (0.25 mL) and CuBr (0.08 g) in dioxane (4.0 mL) for 90 min, and quenched with 1 M AcOH (4.0 mL) according to the method reported by Casara et al.14) The residue obtained from the Et2O extract was passed through a silica gel (7 g) column eluting with benzene–acetone (40 : 1), to afford compound 16a (0.47 g, 63% yield for 3 steps).
N-[3-[2,3-Butadienyl(tert-butoxycarbonyl)amino]propyl]-N,N′-bis(tert-butoxycarbonyl)-1,4-butanediamine (16b)Compound 8b (0.68 g, 1.3 mmol) was treated according to Procedure C to obtain compound 12b (0.47 g, 1.24 mmol). This product was treated using Procedure B to obtain the tri-Boc-protected derivative 15b (0.58 g, 1.2 mmol). A portion of this sample (0.4 g, 0.84 mmol) was similarly subjected to the conversion reaction described above for compound 16a to obtain compound 16b (0.29 g, 65% yield for 3 steps).
N-(3-Aminopropyl)-N′-2,3-butadienyl-1,4-butanediamine (N8-butadienyl Spd) (17)Compound 16a (0.47 g, 0.94 mmol) was treated according to Procedure D to afford the hydrochloride of compound 17 (0.25 g, 87%). 1H-NMR (D2O) δ: 5.14 (1H, q, J=6.7 Hz), 4.86 (2H, d, J=6.7, 2.7 Hz), 3.46 (2H, d, J=6.7, 2.7 Hz), 2.88–3.00 (8H, m), 1.86–1.95 (2H, m), 1.54–1.64 (4H, m). Anal. Calcd for C11H26N3Cl3: C, 43.08; H, 8.54; N, 13.70. Found: C, 42.93; H, 8.40; N, 13.71. Electrospray ionization-time-of-flight (ESI-TOF) MS m/z: 198.1967 (Calcd for C11H23N3+H+: 198.1970).
N-[3-(2,3-Butadienylamino)propyl]-1,4-butanediamine (N1-butadienyl Spd) (18)Compound 16b (0.29 g, 0.59 mmol) was treated according to Procedure D to afford the hydrochloride of compound 18 (0.15 g, 85%). 1H-NMR (D2O) δ: 5.16 (1H, q, J=6.7 Hz), 4.89 (2H, d, J=6.7, 2.6 Hz), 3.50 (2H, d, J=6.7, 2.6 Hz), 2.85–3.05 (8H, m), 1.91–2.01 (2H, m), 1.54–1.68 (4H, m). Anal. Calcd for C11H26N3Cl3: C, 43.08; H, 8.54; N, 13.70. Found: C, 43.12; H, 8.54; N, 13.73. ESI-TOF MS m/z: 198.1965 (Calcd for C11H23N3+H+: 198.1970).
N-(3-Aminopropyl)-N′-2-propenyl-1,4-butanediamine (N8-propenyl Spd) (10)Compound 9a was treated according to Procedure D to afford the hydrochloride of compound 10. 1H-NMR (D2O) δ: 5.72 (1H, ddt, J=17.2, 10.3, 6.8 Hz), 5.32 (1H, dq, J=17.2, 1.2 Hz), 5.30 (1H, dq, J=10.3, 1.2 Hz), 3.48 (2H, br d, J=6.8 Hz), 2.86–3.00 (8H, m), 1.86–1.94 (2H, m), 1.54–1.64 (4H, m). Anal. Calcd for C10H26N3Cl3: C, 40.76; H, 8.89; N, 14.26. Found: C, 40.62; H, 8.65; N, 14.25. ESI-TOF MS m/z: 186.1964 (Calcd for C10H23N3+H+: 186.1970).
N-[3-(2-Propenylamino)propyl]-1,4-butanediamine (N1-propenyl Spd) (11)Compound 9b was treated according to Procedure D to afford the hydrochloride of compound 11. 1H-NMR (D2O) δ: 5.75 (1H, ddt, J=17.1, 10.3, 6.8 Hz), 5.36 (1H, dq, J=17.1, 1.2 Hz), 5.34 (1H, dq, J=10.3, 1.2 Hz), 3.53 (2H, br d, J=6.8 Hz), 2.85–3.02 (8H, m), 1.90–1.99 (2H, m), 1.54–1.67 (4H, m). Anal. Calcd for C10H26N3Cl3: C, 40.76; H, 8.89; N, 14.26. Found: C, 40.91; H, 8.68; N, 14.16. ESI-TOF MS m/z: 186.1964 (Calcd for C10H23N3+H+: 186.1970).
N-(3-Aminopropyl)-N′-2-propynyl-1,4-butanediamine (N8-propynyl Spd) (13)Compound 12a was treated according to Procedure D to afford the hydrochloride of compound 8. 1H-NMR (D2O) δ: 3.75 (2H, m), 2.88–3.06 (8H, m), 2.80 (1H, t, J=2.6 Hz), 1.86–1.95 (2H, m), 1.57–1.65 (4H, m). Anal. Calcd for C10H24N3Cl3: C, 41.04; H, 8.26; N, 14.36. Found: C, 40.81; H, 8.10; N, 14.28. ESI-TOF MS m/z: 184.1809 (Calcd for C10H21N3+H+: 184.1814).
N-[3-(2-Propynylamino)propyl]-1,4-butanediamine (N1-propynyl Spd) (14)Compound 12b was treated according to Procedure D to afford the hydrochloride of compound 14. 1H-NMR (D2O) δ: 3.80 (2H, m), 2.85–3.14 (8H, m), 2.85 (1H, m), 1.92–2.02 (2H, m), 1.54–1.68 (4H, m). Anal. Calcd for C10H24N3Cl3: C, 41.04; H, 8.26; N, 14.36. Found: C, 41.00; H, 8.25; N, 14.29. ESI-TOF MS m/z: 184.1808 (Calcd for C10H21N3+H+: 184.1814).
Enzyme Sources and Assay ProceduresHuman SMO cDNA and hAPAO cDNA were expressed in HEK 293 cells as described previously.5) The cells were cultured for 24 h after transfection, and they were harvested and solubilized with CelLytic M (Sigma-Aldrich). After centrifugation at 20000×g for 15 min, the enzymes were purified from the supernatant by two-steps affinity chromatography using anti-DYKDDDDK antibody beads and anti-c-Myc antibody beads. The purified enzymes were dissolved in 50% glycerol, with 0.09% NaN3 in phosphate buffered saline (PBS)(−). The enzyme solutions were concentrated using Amicon Ultra centrifuge filters, and stored at −20°C.
The standard assay mixture (0.1 mL) consisted of 40 mM borate buffer (pH 9.0) containing 0.5 mg/mL bovine serum albumin (BSA), 0.2 mM 13C2,15N2-spermine for SMO or 0.2 mM 13C2,15N2-N1-acetylspermine for APAO, and the enzyme. After incubation for 30 min at 37°C, an appropriate amount of the internal standard (IS), N-(3-aminopropyl-15N)-1,4-15N-butanediamine-1,1,4,4-d4 (15N3,D4-spermidine), was added to the incubation mixtures, and the mixture was deproteinized with trichloroacetic acid (TCA) (5% final concentration). The supernatant was then applied to a CM-cellulose column (0.3 mL, Wako) and eluted stepwise with pyridine-acetic acid buffer as previously described.5) The polyamine fractions were then reacted with heptafluorobutyric anhydride, and the heptafluorobutyryl derivatives of polyamines were analyzed by mass spectrometry.5)
MS ProceduresThe apparatus used was an Agilent 6530 accurate-mass Q-TOF LC/MS system with a Dual-ESI ion source (Agilent Technologies, CA, U.S.A.). The mobile phase consisted of 0.05% formic acid and 0.2% ammonium acetate in 50% aqueous acetonitrile. The mobile phase flow rate was set to 0.1 mL/min, and the sample injection volume was 1 µL. Standard conditions for Q-TOF MS were used: positive and extended dynamic range mode (2 GHz); Vcap, 3500 V; nebulizer pressure, 35 psi; drying gas, 8 L/min; gas temp, 300°C; fragmentor, 150 V; skimmer, 65 V; Oct 1 RF Vpp, 750 V. Accurate mass spectra were recorded across a range of m/z 100 to 1100. The observed ions for the heptafluorobutyryl derivatives of spermidine, 13C2,15N2-spermidine and 15N3,D4-spermidine (IS) were m/z 751, 755, and 758, respectively, represented as [M+NH4]+. The determinations were based on the peak area ratio of Spd or 13C2,15N2-Spd to 15N3,D4-Spd (IS).