Abstract
Red ginseng has long been used as a traditional medicine in many East Asian countries including Korea. It is known to exhibit various pharmacological effects, including anti-oxidant, anti-cancer, anti-stress and anti-diabetes activities. To further explore its actions, the present study evaluated effects of Korean red ginseng (KRG) extract on neuronal injury induced by various types of insults using primary cultured rat cortical cells. KRG extract inhibited neuronal damage and generation of intracellular reactive oxygen species (ROS) induced by excitatory amino acids, such as glutamate and N-methyl-D-aspartate (NMDA), or by Aβ(25–35). To elucidate possible mechanism(s) by which KRG extract exerts neuroprotective action, its effects on apoptosis and apoptosis-related signaling molecules in neurons were assessed. KRG extract markedly increased phosphorylation of Bad at Ser 112 and inhibited Bax expression and caspase 3 activity. It also inhibited DNA fragmentation induced by NMDA or Aβ(25–35). These results indicate that KRG extract protects cultured neurons from excitotoxicity and Aβ(25–35)-induced toxicity through inhibition of ROS generation and apoptotic cell death. In addition, KRG extract inhibited β-secretase activity, implying that it may reduce Aβ peptide formation. Taken together, these findings suggest that KRG extract may be beneficial for the prevention and/or treatment of neurodegenerative disorders including Alzheimer’s disease.
Glutamate is an excitatory amino acid neurotransmitter in the mammalian brain. Glutamate-induced neurotransmission is mediated through ionotropic glutamate receptor subtypes, such as N-methyl-D-aspartic acid (NMDA), α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid and kainate receptors.1) Excess release of glutamate has been implicated in many neurological disorders including Alzheimer’s disease (AD) and cerebral ischemia.2,3) It has been revealed that overactivation of NMDA receptor causes excess influx of calcium ions into cells, which can trigger mitochondrial membrane depolarization, free radical generation, lipid peroxidation, caspase activation and cellular toxicity. These events ultimately induce apoptosis or necrosis which eventually causes cellular dysfunction and death.4,5) Therefore, pharmacological interventions regulating excessive excitatory transmission may be beneficial for preventing or treating neurodegenerative diseases such as AD. For example, memantine, a non-competitive NMDA receptor antagonist, blocks excessive excitatory transmission and is clinically used in the treatment of AD.6)
AD is a progressive neurodegenerative disorder characterized by cognitive deficits and memory loss.7) One of the main pathological features of AD is the appearance of senile plaques formed by the extracellular deposition of β-amyloid (Aβ) peptide. This peptide is derived from amyloid precursor protein (APP) following the enzymatic cleavage by β- and γ-secretases.8) Aβ peptide appears to be an important instigator of cellular abnormalities. More specifically, it causes impairment of energy metabolism, destabilization of calcium homeostasis and induction of apoptosis, ultimately leading to neuronal cell dysfunction and death.9) In addition, Aβ peptide induces the oxidative damage of cells through the generation of reactive oxygen species (ROS) which may initiate neurotoxic events. Moreover, it has been suggested that an increase in ROS and free radicals generates more Aβ peptide, which in turn imposes further oxidative damage and toxic insults on the cells.10) Accordingly, inhibition of neuronal cell damage caused by Aβ peptide or reduction of its production may be considered as promising approaches for the prevention and treatment of AD.11,12) Although a number of studies have addressed these approaches to combat this disease, to date no fruitful outcome has been yet achieved.
The use of traditional herbs including Panax ginseng C. A. Meyer (Araliaceae) in the treatment of many diseases has been reported by many researchers.13) Red ginseng is made of ginseng that has been cultivated for 4–6 years and subjected to repeated steaming and drying processes. The extract obtained from Korean red ginseng (KRG) has been reported to exhibit various pharmacological actions, including anti-oxidant, anti-cancer, anti-stress and anti-diabetes activities.14–17) Additionally, a number of studies have shown the effects of KRG extract on the central nervous system (CNS). It has been reported in vitro and in vivo that KRG extract attenuates kainate-induced excitotoxicity by antioxidative effects.18) It also exerts anti-apoptotic action against H2O2-induced oxidative stress in SK-N-SH cells by decreasing caspase 3 activity and increasing Bcl-2 expression.19) Moreover, it has been shown that KRG extract improves scopolamine-induced memory impairment in mice.20) This information raises considerable interest on the possible use of KRG extract for the prevention and treatment of neurodegenerative disorders including AD.
In an attempt to expand and characterize the pharmacological actions of KRG extract in the CNS, the present study evaluated its effects on neuronal cell damage induced by various types of insults including excitatory amino acids and Aβ using primary cultured rat cortical cells. We also investigated the possible mechanism(s) by which KRG extract exerts neuroprotective effects by evaluating its impact on apoptosis and apoptosis-related molecules in cells. Furthermore, we also tested its effect on β-secretase, an enzyme involved in the Aβ formation from APP.
MATERIALS AND METHODS
MaterialsMinimum essential medium (MEM), fetal bovine serum (FBS), horse serum (HS), and antibiotic-antimycotic agent were purchased from Gibco BRL (Grand Island, NY, U.S.A.). Laminin, cytosine arabinoside, L-glutamic acid and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were procured from Sigma-Aldrich (St. Louis, MO, U.S.A.). NMDA was obtained from Tocris Bioscience (Bristol, U.K.) and Aβ(25–35) was purchased from Bachem Ltd. (San Carlos, U.K.). All other chemicals were of analytical grade. Anti-phospho-Bad (Ser 112), anti-Bad, anti-Bax, anti-caspase 3 and anti-poly(ADP-ribose) polymerase (PARP) antibodies as well as horseradish peroxidase (HRP)-conjugated anti-rabbit immunoglobulin G (IgG) and anti-mouse IgG were purchased from Cell Signaling Technology (Danvers, MA, U.S.A.).
Timed-pregnant Sprague-Dawley (SD) rats were purchased from Daehan Biolink (Chungbuk, Korea). The animals were maintained in an animal room under a controlled temperature (22±2°C) with a 12 h light-dark cycle. They were given access to a standard chow diet and water ad libitum. All experimental steps, including the care and handling of the animals, were conducted following the international guidelines (Guide for the Care and Use of Laboratory Animals, Institute of Laboratory Animal Resources, Commission on Life Sciences, National Research Council, U.S.A.; National Academy Press: Washington D.C., 1996).
Preparation of KRG ExtractKRG extract was manufactured and supplied by Korea Ginseng Corporation (Seoul, Korea) from the roots of 6-year-old fresh ginseng (Panax ginseng Meyer) harvested in Korea. These roots were steamed for 3 h at 90–100°C and dried at 50–80°C. KRG extract was then prepared from KRG water extract obtained through decoction of the roots three times at 85–90°C for 8 h using a circulating hot water bath. The water content of the pooled extract was 36% of the total weight. Prior to the experimental use, KRG extract was diluted appropriately with sterile phosphate buffered saline (PBS), pH 7.4, to produce the working stock solution.
Preparation of Aggregated Aβ(25–35)Aggregated Aβ(25–35) was prepared as previously described.21) In brief, Aβ(25–35) was dissolved to 1 mM with sterile distilled water and incubated for 7 d at 37°C.
Primary Cultures of Rat Cortical CellsPrimary cultures of cortical cells containing neuronal and non-neuronal cells were prepared from the brains of SD rat embryos at 17 d gestation as described previously.22–24) In brief, cerebral cortices of the embryos were dissected and mechanically dissociated into single cells by trituration using fire-polished Pasteur pipettes. The isolated cells were seeded in MEM supplemented with 5% FBS, 5% HS and 1% antibiotic-antimycotic agent at a density of 1–6×105 cells/well of a 24- or 96-well culture plate (BD Falcon, Franklin Lakes, NJ, U.S.A.) or at a density of 6×106 cells per a 35 mm culture dish (Corning, NY, U.S.A.) pre-coated with laminin and poly-L-lysine. The cells were maintained in an incubator at 37°C with a humidified atmosphere of 95% air/5% CO2. Proliferation of non-neuronal cells in the culture was arrested by the treatment with 10 µM cytosine arabinoside at 7 d after seeding of cells. All experiments were carried out at 10–11 d after plating.
Exposure of Cultured Cells to Various Types of InsultsCells were washed with N-(2-hydroxyethyl)piperazine-N′-2-ethanesulfonic acid (HEPES)-buffered control salt solution (HCSS, 120 mM NaCl, 5.4 mM KCl, 1.6 mM MgCl2·6H2O, 2.3 mM CaCl2·2H2O, 15 mM glucose, 10 mM NaOH, 20 mM HEPES, pH 7.4), and excitotoxic neuronal damage was induced by the exposure of cultured cells to 100 µM L-glutamate in HCSS or 300 µM NMDA in Mg2+-free HCSS for 15 min.23–25) The control cells were treated with HCSS or Mg2+-free HCSS without any agent. The cells were then washed with HCSS and maintained in MEM for 22–24 h in the incubator. To induce neurotoxicity by Aβ, cells were exposed to aggregated Aβ(25–35) at a final concentration of 40 µM in MEM for 24 h at 37°C.24) The control cells were treated with MEM without any agent.
To investigate effects of KRG extract on the neuronal damage induced by glutamate, NMDA or Aβ(25–35), the cells were simultaneously treated with various concentrations of KRG extract in combination with the respective insults as mentioned above. Following the desired treatments, the viability of cells was assessed as described below.
Assessment of Cell ViabilityFollowing the desired treatments, the cell viability was determined using MTT reduction assay.24) Briefly, MTT was added to the treated cells at a final concentration of 1 mg/mL in PBS and the cells were incubated for 3 h at 37°C. The culture media were carefully removed from the wells and dimethylsulfoxide was added to the cells to dissolve the formazan crystal products. The absorbance was read at 550 nm using a microplate reader (SpectraMax M2e, Molecular Devices, Sunnyvale, CA, U.S.A.). The cell viability was expressed as percentage of control-treated cells.
Measurement of Intracellular ROSSpectrofluorometric determination of intracellular ROS levels was performed using 2′,7′-dichlorofluorescin diacetate (DCFH-DA, Sigma-Aldrich) as a fluorescent probe.26,27) Briefly, cells were incubated with 10 µM DCFH-DA for 30 min at 37°C to allow the diffusion of the fluorescent probe into the cells and its subsequent hydrolysis to non-fluorescent dichlorofluorescin (DCFH) under the action of intracellular esterases. After removal of excess DCFH-DA, cells were washed with HCSS and then exposed to either L-glutamate or NMDA for 2 h at 37°C in the presence or absence of various concentrations of KRG extract. In other experiments, cells were exposed to aggregated Aβ(25–35) at a final concentration of 40 µM in MEM for 24 h at 37°C in the presence or absence of various concentrations of KRG extract. The treated cells were then washed with HCSS and incubated with 10 µM DCFH-DA for 30 min at 37°C. Intracellular production of ROS was measured by the fluorescence detection of dichlorofluorescein (DCF) as the oxidized product of DCFH on a microplate reader (SpectraMax M2e, Molecular Devices) with an excitation wavelength of 490 nm and emission wavelength of 520 nm. ROS generation was quantitated as the fluorescence intensity of DCF.
Western BlottingFollowing the treatments, the cells were washed with cold PBS and lysed in the lysis buffer (10 mM Tris–HCl, pH 7.5, 150 mM NaCl, 2 mM ethylenediaminetetraacetic acid (EDTA), 4.5 mM sodium pyrophosphate, 10 mM β-glycerophosphate, 1 mM NaF, 1 mM Na3VO4, 1% (v/v) Triton X-100, 0.5% Nonidet P-40 and one tablet of protease inhibitor cocktail (Roche Diagnostic GmbH, Mannheim, Germany) per 10 mL of lysis buffer) for 30 min on ice. Lysates were centrifuged at 14000 rpm for 30 min at 4°C to remove insoluble materials and the supernatant was stored at −80°C until used. The protein concentrations of the supernatants were determined using a DC™ protein assay kit (Bio-Rad, Hercules, CA, U.S.A.). After being denatured at 100°C in Laemmli sample buffer containing 100 mM dithiothreitol, equal amount of lysate proteins were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) on either 8% or 12% gels.28) Resolved proteins were electrophoretically transferred to nitrocellulose membrane (Schleicher and Schnell Bioscience, Dassel, Germany) for 1.5 h at 100 V. The membranes were blocked with 5% skim milk for 1.5 h at room temperature and incubated overnight at 4°C with the specific primary antibodies as mentioned in the figure legends. After washing, the membranes were incubated for 1.5 h with appropriate HRP-conjugated anti-IgG secondary antibodies, and the immunoreactive bands were detected by Bio-Rad ChemiDoc XRS imaging system using SuperSignal West Pico ECL reagent (Thermo Fisher Scientific, Rockford, IL, U.S.A.).
Terminal Deoxynucleotidyl Transferase-Mediated Deoxyuridine Triphosphate (dUTP) Nick-End Labeling (TUNEL) AssayThe apoptosis of cells was determined using a TUNEL assay kit (DeadEnd™ Colorimetric TUNEL System Promega, Madison, WI, U.S.A.) according to the manufacturer’s instructions. Briefly, following the desired treatments, the cells were washed with PBS and fixed in 4% paraformaldehyde for 25 min. The cells were then washed with PBS and permeabilized with 0.2% Triton X-100 for 5 min. Following this, the cells were washed with PBS and equilibrated with equilibration buffer for 10 min at room temperature. After this, the cells were incubated in terminal deoxynucleotidyl transferase reaction mixture containing biotinylated nucleotide mix for 60 min at 37°C to allow the end-labeling reaction. The cells were then immersed in 2X saline sodium citrate solution for 15 min, and subsequently incubated with streptavidin-conjugated HRP for 30 min at room temperature. The cells were then washed with PBS and finally stained with diaminobenzidine. Stained TUNEL-positive cells were confirmed using a TS-100 inverted microscope (Nikon, Tokyo, Japan) at 400× magnification.
Assay of β-Secretase ActivityThe effect of KRG extract on the enzymatic activity of β-secretase was determined using a β-secretase FRET assay kit (PanVera, Madison, WI, U.S.A.) according to the manufacturer’s instructions with some modification. Briefly, a 10 µL aliquot of assay buffer containing increasing concentration of KRG extract was mixed with 20 µL of the substrate (750 nM) in individual wells of a 96-well plate. To this mixture, 10 µL of β-secretase enzyme (1.0 unit/mL) was added and mixed well. This mixture was reacted at room temperature for 2 h and then the fluorescence measurement was carried out on a microplate reader (SpectraMax M2e, Molecular Devices) with excitation and emission wavelengths set at 545 and 585 nm, respectively.
Statistical AnalysisAll experiments were conducted at least three times. Quantitative data are expressed as the mean±S.E.M. Statistical analysis was performed by one-way ANOVA followed by Tukey’s post hoc test using Sigma Stat 3.5 software (Systat Software, Point Richmond, CA, U.S.A.). A p<0.05 were considered statistically significant.
RESULTS
Effect of KRG Extract on the Cell Viability in Primary Cultured Rat Cortical CellsIn order to evaluate if KRG extract exhibits cytotoxicity in primary cultured rat cortical cells, we have examined the viability of cells after exposure to KRG extract at concentrations ranging from 0.1–10 mg/mL for the time periods of 6, 12 and 24 h. As can be seen in Fig. 1, KRG extract did not elicit any cytotoxicity at any time periods of exposure at the concentrations up to 3 mg/mL. However, it significantly reduced the viability of cells at 10 mg/mL in a time-dependent manner (Fig. 1). Based on these findings, the remaining experiments were performed using the concentrations of KRG extract below 10 mg/mL.
Effects of KRG Extract on Excitotoxic Neuronal Cell DamageKRG Extract Inhibits Glutamate- or NMDA-Induced Excitotoxicity and ROS GenerationAs reported previously, primary cultured rat cortical cells are vulnerable to glutamate- or NMDA-induced excitotoxicity.23–25) In this study, we confirmed these findings using the MTT reduction assay, showing that the viability of the cultured cortical cells was reduced by approximately 30–40% as compared to the control cells upon exposure to either L-glutamate or NMDA (Figs. 2A, B). To evaluate the protective effect of KRG extract on the glutamate- or NMDA-induced excitotoxic cell damage, the cultured cells were simultaneously treated with various concentrations of KRG extract. KRG extract provided a marked protection against the excitotoxic damage induced by the two excitatory amino acids (Figs. 2A, B). At the concentration of 3 mg/mL, KRG extract almost completely rescued the cells from the excitotoxic injury, exhibiting the cell viability similar to control cells. We also assessed the effect of memantine on the excitotoxic injury for reference in this study. The protective effects of KRG extract at 3 mg/mL were even more pronounced than that of memantine (Figs. 2A, B).
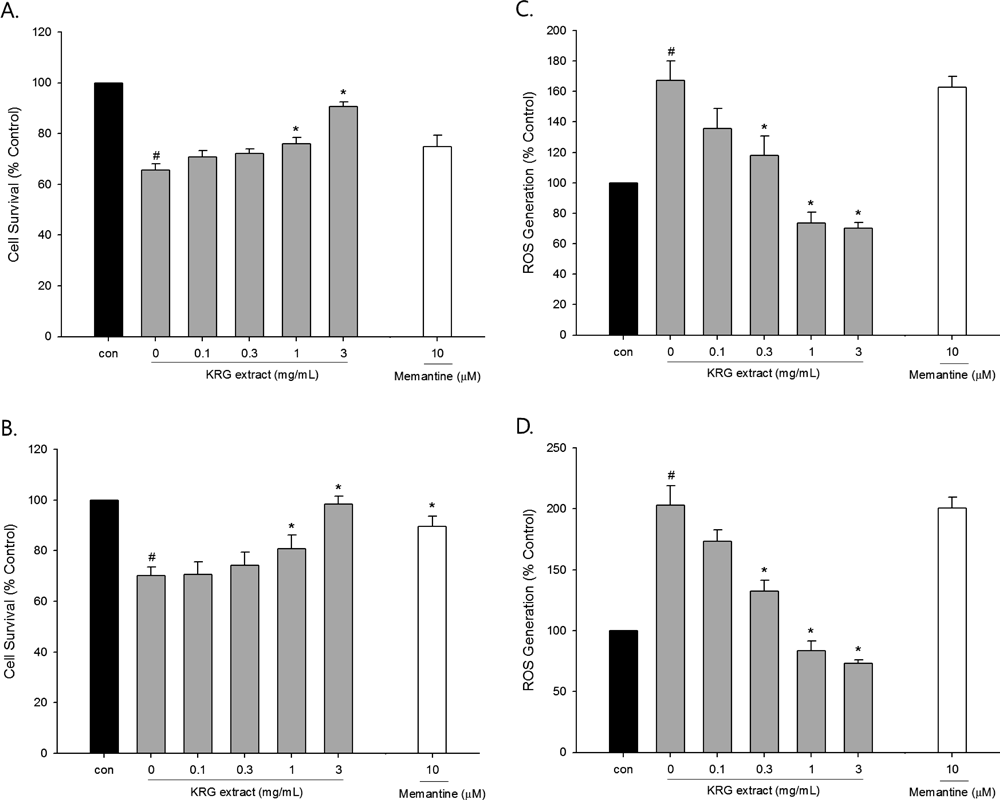
To understand the possible mechanism(s) by which the KRG extract protected against the excitotoxic damage, we next evaluated its effects on glutamate- or NMDA-induced intracellular ROS generation. Treatment of cultured cortical cells with glutamate or NMDA significantly augmented the intracellular ROS level (1.7–2-fold enhancement over control; Figs. 2C, 2D). The glutamate- or NMDA-induced intracellular ROS generation was inhibited in a concentration-dependent manner by co-treatment of cells with KRG extract, with the maximal effect achieved at 1 and 3 mg/mL. However, memantine did not inhibit the glutamate- or NMDA-induced intracellular ROS generation at the concentration tested in this study.
KRG Extract Protects from Glutamate or NMDA-Induced Excitotoxic Cell Damage through the Inhibition of ApoptosisTo further elucidate the possible action mechanisms, we evaluated a number of molecular events that play key roles in apoptotic signaling pathway, such as phosphorylation of Bad, expression of Bax and cleavage of caspase 3. Phosphorylation of Bad at Ser 112 is known to inhibit apoptotic activity of Bad protein.29) Our findings have revealed that the phosphorylation of Bad at this position was markedly increased in glutamate- or NMDA-treated cells being co-exposed to KRG extract (Fig. 3A). Treatment with either glutamate or NMDA produced marked increases in Bax expression and cleaved caspase 3. The NMDA-induced Bax expression and cleaved caspase 3 were dramatically inhibited by KRG extract (Fig. 3A). Similarly, the glutamate-induced cleavage of caspase 3 was markedly inhibited by KRG extract, while the enhanced Bax expression was only minimally affected (Fig. 3A).
We then confirmed the anti-apoptotic action of KRG extract by evaluating its effect on DNA fragmentation, another hallmark of apoptosis, using the TUNEL assay. Compared to the control-treated cells (Fig. 3B, a), exposure of the cultured cells to NMDA caused substantial excitotoxic cell damage with the appearance of a large number of TUNEL-positive cells (Fig. 3B, b). Co-exposure of NMDA-treated cells to KRG extract at the concentration of 3 mg/mL rescued the cells from the excitotoxic injury, with a marked reduction of the apoptotic cell population (Fig. 3B, c). Treatment with KRG extract alone appeared not to cause cell damage or DNA fragmentation at this concentration (Fig. 3B, d).
Effects of KRG Extract on Aβ(25–35)-Induced Neuronal Cell DamageKRG Extract Inhibits Aβ(25–35)-Induced Neuronal Damage and ROS GenerationTo further characterize pharmacological actions of KRG extract in cultured neurons, we extended our study to examine the effects of this extract on the neuronal cell damage caused by exposure to Aβ(25–35), the active fragment of Aβ peptide. The viability of the cells exposed to 40 µM Aβ(25–35) for 24 h was significantly reduced to 65%, as compared to control-treated cells (Fig. 4A). KRG extract provided appreciable protection against the Aβ-induced neurotoxicity, resulting in significantly improved cell viability at 0.3 and 1 mg/mL.
We also evaluated the effects of KRG extract on Aβ-induced intracellular ROS generation using DCFH-DA as a fluorescent probe. As shown in Fig. 4B, exposure of cells to Aβ(25–35) increased intracellular ROS levels, which was significantly inhibited by the co-treatment with KRG extract at 0.3 and 1 mg/mL. In contrast, memantine had no effect on the Aβ(25–35)-induced injury or ROS at the concentration tested (data not shown).
KRG Extract Protects from Aβ(25–35)-Induced Neuronal Damage through the Inhibition of ApoptosisWe then evaluated the effects of KRG extract on the Aβ-induced alterations of apoptotic signaling molecules including Bad, Bax, caspase 3 and PARP proteins. The phosphorylation of Bad at Ser 112 was markedly increased by KRG extract (Fig. 5A). In addition, KRG extract almost completely blocked the Aβ-induced cleavages of caspase 3 and PARP (Fig. 5A). However, the enhanced Bax expression in Aβ-treated cells was only slightly inhibited by KRG extract (Fig. 5A).

Again, we confirmed the anti-apoptotic action of KRG extract using TUNEL assay. Similar to the findings in the cells treated with NMDA in the absence of KRG extract (Fig. 3B, b), a considerably large number of TUNEL-positive cells was evident in the culture exposed to Aβ(25–35) for 18 h (Figs. 5B, b, C, b). Co-exposure of Aβ-treated cells to KRG extract at either 0.3 or 3 mg/mL resulted in a marked decrease in the population of TUNEL-positive cells (Figs. 5B, c, C, c, respectively).
Effect of KRG Extract on β-Secretase ActivityTo explore additional pharmacological properties of KRG extract, we tested its effect on β-secretase activity, the enzyme involved in the production of Aβ peptide from APP.8) We found that KRG extract concentration-dependently inhibited β-secretase activity (Fig. 6), implying that it may reduce the Aβ peptide formation.
DISCUSSION
Panax ginseng C. A. Mayer (Araliceae) has been used as a traditional herbal medicine in many oriental countries.30) KRG made by steaming and drying process of ginseng is known to gain some beneficial properties through the chemical transformation of active components of ginseng during the steaming process.31) The extract obtained from KRG has been reported to exhibit a variety of pharmacological actions, suggesting that it may be beneficial for several types of disorders including cancer and diabetes.14–17) In addition, KRG extract has been shown to protect SK-N-SH cells19) and hippocampal cells18) against H2O2-induced oxidative stress and kainate-induced neurotoxicity, respectively, implying its additional beneficial actions in the CNS. We observed in this study that KRG extract inhibited glutamate- or NMDA-induced excitotoxicity as well as Aβ-induced neurotoxicity in primary cultured rat cortical cells. Based on our findings, the neuroprotective action exerted by KRG extract is associated with inhibition of ROS generation and apoptotic cell death caused by these insults.
Excessive release of glutamate is one of the crucial causes for cellular dysfunction and death in many neurodegenerative disorders including AD. Overactivation of glutamatergic receptors by brief and intense exposure to glutamic acid triggers a variety of neurotoxic events, which ultimately leads to excitotoxic cell death.5) Based on our MTT assay, treatment of the primary cultured rat cortical cells with 100 mM of glutamate for 15 min resulted in approximately 40% cell death over the subsequent 22–24 h (Fig. 2A), which is in agreement with previous observations.23,24) We tested the effect of KRG extract on the glutamate-induced excitotoxicity at the concentrations of 0.1–3 mg/mL, since it did not elicit any toxicity at these concentrations even in the cells treated for 24 h (Fig. 1). The glutamate-induced cell death was significantly inhibited by KRG extract at 1 and 3 mg/mL (Fig. 3A). Exposure of the cultured cells to glutamic acid increased intracellular ROS production up to 1.7-fold that of control-treated cells, as measured using the fluorescent probe DCFH-DA (Fig. 2C). The glutamate-induced ROS generation was also inhibited by KRG extract.
Among the receptors involved in the glutamate-mediated excitotoxicity, NMDA receptor has crucial roles in triggering acute processes of cell dysfunction.32) Thus, we next examined whether KRG extract inhibited the excitotoxicity and ROS generation induced by NMDA exposure. As shown in Figs. 2B and D, the NMDA-induced excitotoxicity and ROS generation were significantly inhibited by KRG extract. These findings demonstrate that KRG extract protects cultured neurons from excitotoxic damage, and that inhibition of ROS generation may contribute to its neuroprotective action.
Memantine, a non-competitive NMDA receptor antagonist, is a drug clinically used for the treatment of AD patients. We assessed the effect of memantine and compared it to that of KRG extract. Prior to our study, we have conducted preliminary experiments to optimize the concentration of memantine. In our preliminary experiments, effects of memantine were tested on the NMDA-induced excitotoxicity at the concentrations ranging from 0.3 to 30 µM (data not shown). We found that this drug at 10 µM inhibited the NMDA-induced excitotoxicity with submaximal efficacy, as shown in Fig. 2B. Moreover, previous studies have estimated that 10 µM concentration of memantine is within the range available to NMDA receptors in cellular environment after administration of a clinical dose of the compound.33–35) Based on these, we have selected the concentration of memantine at 10 µM for the evaluation of this drug as a reference in the present study.
As expected, memantine inhibited the glutamate- and NMDA-induced excitotoxicity (Figs. 2A, B). Notably, the inhibition achieved by memantine at the concentration tested in this study (10 µM) was almost comparable to that of KRG extract at 1 mg/mL. Interestingly, KRG extract at 3 mg/mL exhibited a more pronounced neuroprotective effect than memantine, increasing cell survival almost up to the control levels (Figs. 2A, B). In contrast, memantine showed no effect on the glutamate- or NMDA-induced ROS production (Figs. 2C, D), implying that the action mechanism involved in the pharmacological actions of memantine may be different from that of KRG extract.
Substantial evidence has shown that the excitotoxicity induced by glutamate or NMDA is associated with apoptotic and/or necrotic cell death.4) Thus, we evaluated its effects on apoptosis and apoptosis-related molecules after inducing excitotoxicity in the cultured cells.
Bcl-2 family proteins as key regulators of apoptosis include anti-apoptotic proteins such as Bcl-2 and Bcl-xL, and pro-apoptotic proteins such as Bax, Bak, Bad, Bid and Bim.36) Bad interacts with anti-apoptotic molecules Bcl-2 and Bcl-xL and promotes apoptosis. On the other hand, phosphorylated Bad at Ser 112 and 136 prevents heterodimer formation with Bcl-2 or Bcl-xL, which promotes cell survival.29,37) Bax is a pro-apoptotic Bcl-2 family member that exists as a monomer in the cytosol. In response to apoptotic signals, it interacts with BH3-only proteins including Bad, Bid and Bim and induces conformational change, which leads to pore formation of mitochondria and releases cytochrome c and other apoptotic proteins.38–41) Cytochrome c released from the mitochondria works in combination with other proteins, such as caspase 9 and apoptosis-activating factor 1 (Apaf-1), to process procaspase-3.42,43) As an executioner caspase, the procaspase-3 is cleaved and activated by an initiator caspase after apoptotic signaling events have occurred.44) The cleaved caspase 3 leads to DNA fragmentation, a key feature of apoptosis, by inactivating inhibitor of caspase activated DNase (ICAD).45)
Evaluation of KRG extract on these apoptosis-related molecules under excitotoxic condition of the cultured cells showed that it increased phosphorylation of Bad at Ser 112, and inhibited Bax expression and caspase 3 activation (Fig. 3A). Furthermore, KRG extract inhibited NMDA-induced DNA fragmentation as assessed by the TUNEL assay (Fig. 3B). These results suggest that inhibition of apoptotic signal is one of the important mechanisms by which KRG extract protects against excitotoxicity.
Among the diverse pathophysiological factors associated with AD,7,10) Aβ peptide is one of the main features to cause toxicity leading to neuronal cell death.9) Exposure of the cultured cells to Aβ(25–35), an active fragment of Aβ peptide, produced significant reduction of cell survival up to 65% of control (Fig. 4A). KRG extract significantly increased the cell survival at the concentrations of 0.3 and 1 mg/mL. The ROS generated by Aβ treatment was also inhibited by KRG extract (Fig. 4B). Additionally, this extract increased the phosphorylation of Bad at Ser 112, and markedly inhibited caspase 3 activation and DNA fragmentation induced by the exposure to Aβ (Figs. 5A–C). Interestingly, exposure of cells to Aβ markedly increased cleavage of PARP, which was dramatically inhibited by KRG extract (Fig. 5A). PARP, another important protein for cell survival, is one of the main cleavage targets of caspase 3 during apoptosis.46) The cultured cells exposed to either glutamate or NMDA did not show PARP cleavage (data not shown), implying that different apoptotic pathways may be triggered during excitotoxicity. Collectively, our results indicate that inhibition of Aβ-induced apoptotic signals including PARP cleavage might be a contributing factor for the neuroprotective action exerted by KRG extract.
In addition to its neuroprotective actions, our data from β-secretase FRET assay imply that KRG extract may additionally reduce Aβ peptide formation by inhibiting β-secretase activity (Fig. 6).
Taken together, these results indicate that KRG extract protects primary cultured rat cortical cells from excitotoxicity and Aβ-induced neuronal cell damage, and that inhibition of ROS generation as well as apoptotic cell death caused by the respective insults is involved in its neuroprotective action. Reduction of Aβ formation by KRG extract along with its neuroprotective actions may exert beneficial influence for AD pathology. Further study is necessary to elucidate the active principle(s) in KRG extract exhibiting these effects.
Acknowledgment
This research was supported by the Grant (2011–2012) from the Korean Society of Ginseng funded by Korean Ginseng Corporation and the GRRC program of Gyeonggi province [GRRC DONGGUK 2012-B01 & 2013-B01], Republic of Korea.
REFERENCES
- 1) Doble A. The role of excitotoxicity in neurodegenerative disease: implications for therapy. Pharmacol. Ther., 81, 163–221 (1999).
- 2) Meldrum BS. Glutamate as a neurotransmitter in the brain: review of physiology and pathology. J. Nutr., 130 (Suppl.), 1007S–1015S (2000).
- 3) Supnet C, Bezprozvanny I. The dysregulation of intracellular calcium in Alzheimer’s disease. Cell Calcium, 47, 183–189 (2010).
- 4) Martin LJ, Al-Abdulla NA, Brambrink AM, Kirsch JR, Sieber FE, Portera-Cailliau C. Neurodegeneration in excitotoxicity, global cerebral ischemia, and target deprivation: a perspective on the contributions of apoptosis and necrosis. Brain Res. Bull., 46, 281–309 (1998).
- 5) Dong XX, Wang Y, Qin ZH. Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol. Sin., 30, 379–387 (2009).
- 6) Lipton SA. The molecular basis of memantine action in Alzheimer’s disease and other neurologic disorders: low-affinity, uncompetitive antagonism. Curr. Alzheimer Res., 2, 155–165 (2005).
- 7) Lleó A, Greenberg SM, Growdon JH. Current pharmacotherapy for Alzheimer’s disease. Annu. Rev. Med., 57, 513–533 (2006).
- 8) Findeis MA. The role of amyloid beta peptide 42 in Alzheimer’s disease. Pharmacol. Ther., 116, 266–286 (2007).
- 9) Mattson MP. Pathways towards and away from Alzheimer’s disease. Nature, 430, 631–639 (2004).
- 10) Markesbery WR. Oxidative stress hypothesis in Alzheimer’s disease. Free Radic. Biol. Med., 23, 134–147 (1997).
- 11) Sun X, Jin L, Ling P. Review of drugs for Alzheimer’s disease. Drug Discov. Ther., 6, 285–290 (2012).
- 12) Citron M. β-Secretase inhibition for the treatment of Alzheimer’s disease—promise and challenge. Trends Pharmacol. Sci., 25, 92–97 (2004).
- 13) Kim S-K, Park J-H. Trends in ginseng research in 2010. J. Ginseng. Res., 35, 389–398 (2011).
- 14) Kim YK, Guo Q, Packer L. Free radical scavenging activity of red ginseng aqueous extracts. Toxicology, 172, 149–156 (2002).
- 15) Helms S. Cancer prevention and therapeutics: Panax ginseng. Altern. Med. Rev., 9, 259–274 (2004).
- 16) Kaneko H, Nakanishi K. Proof of the mysterious efficacy of ginseng: basic and clinical trials: clinical effects of medical ginseng, korean red ginseng: specifically, its anti-stress action for prevention of disease. J. Pharmacol. Sci., 95, 158–162 (2004).
- 17) Vuksan V, Sievenpipper J, Jovanovski E, Jenkins AL. Current clinical evidence for Korean red ginseng in management of diabetes and vascular disease: a Toronto’s ginseng clinical testing program. J. Ginseng. Res., 34, 264–273 (2010).
- 18) Han JY, Ahn SY, Oh EH, Nam SY, Hong JT, Oh KW, Lee MK. Red ginseng extract attenuates kainate-induced excitotoxicity by antioxidative effects. Evid. Based Complement. Alternat. Med., 2012, 1–10 (2012).
- 19) Kim E-H, Lee M-J, Kim I-H, Pyo S-N, Choi K-T, Rhee D-K. Anti-apoptotic effects of red ginseng on oxidative stress induced by hydrogen peroxide in SK-N-SH cells. J. Ginseng. Res., 34, 138–144 (2010).
- 20) Lee M-R, Yun B-S, Liu L, Zhang D-L, Wang Z, Wang C-L, Gu L-J, Wang C-Y, Mo E-K, Sung C-K. Effect of black ginseng on memory improvement in the amnesic mice induced by scopolamine. J. Ginseng. Res., 34, 51–58 (2010).
- 21) Liu X, Xu K, Yan M, Wang Y, Zheng X. Protective effects of galantamine against Abeta-induced PC12 cell apoptosis by preventing mitochondrial dysfunction and endoplasmic reticulum stress. Neurochem. Int., 57, 588–599 (2010).
- 22) Cho J, Kong J-Y, Jeong D-Y, Lee KD, Lee D-U, Kang B-S. NMDA recepter-mediated neuroprotection by essential oils from the rhizomes of Acorus gramineus. Life Sci., 68, 1567–1573 (2001).
- 23) Cho J, Joo NE, Kong J-Y, Jeong D-Y, Lee KD, Kang B-S. Inhibition of excitotoxic neuronal death by methanol extract of Acori graminei rhizoma in cultured rat cortical neurons. J. Ethnopharmacol., 73, 31–37 (2000).
- 24) Cho J, Kim HM, Ryu JH, Jeong YS, Lee YS, Jin C. Neuroprotective and antioxidant effects of the ethyl acetate fraction prepared from Tussilago farfara L. Biol. Pharm. Bull., 28, 455–460 (2005).
- 25) Choi DW. Excitotoxic cell death. J. Neurobiol., 23, 1261–1276 (1992).
- 26) Rota C, Chignell CF, Mason RP. Evidence for free radical formation during the oxidation of 2′,7′-dichlorofluorescin to the fluorescent dye 2′,7′-dichlorofluorescein by horseradish peroxidase: possible implications for oxidative stress measurements. Free Radic. Biol. Med., 27, 873–881 (1999).
- 27) Moongkarndi P, Srisawat C, Saetun P, Jantaravinid J, Peerapittayamongkol C, Soi-ampornkul R, Junnu S, Sinchaikul S, Chen ST, Charoensilp P, Thongboonkerd V, Neungton N. Protective effect of mangosteen extract against β-amyloid-induced cytotoxicity, oxidative stress and altered proteome in SK-N-SH cells. J. Proteome Res., 9, 2076–2086 (2010).
- 28) Cho J, Gruol DL. The chemokine CCL2 activates p38 mitogen-activated protein kinase pathway in cultured rat hippocampal cells. J. Neuroimmunol., 199, 94–103 (2008).
- 29) Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell, 91, 231–241 (1997).
- 30) Ernst E. Panax ginseng: an overview of the clinical evidence. J. Ginseng. Res., 34, 259–263 (2010).
- 31) Kwon SW, Han SB, Park IH, Kim JM, Park MK, Park JH. Liquid chromatographic determination of less polar ginsenosides in processed ginseng. J. Chromatogr. A, 921, 335–339 (2001).
- 32) Doble A. Excitatory amino acid receptors and neurodegeneration. Therapie, 50, 319–337 (1995).
- 33) Hesselink MB, De Boer BG, Breimer DD, Danysz W. Brain penetration and in vivo recovery of NMDA receptor antagonists amantadine and memantine: a quantitative microdialysis study. Pharm. Res., 16, 637–642 (1999).
- 34) Danysz W, Parsons CG, Mobius HJ, Stoffler A, Quack G. Neuroprotective and symptomatological action of memantine relevant for Alzheimer’s disease—a unified glutamatergic hypothesis on the mechanism of action. Neurotox. Res., 2, 85–97 (2000).
- 35) Wild AR, Akyol E, Brothwell SL, Kimkool P, Skepper JN, Gibb AJ, Jones S. Memantine block depends on agonist presentation at the NMDA receptor in substantia nigra pars compacta dopamine neurones. Neuropharmacology, 73, 138–146 (2013).
- 36) Kim H, Rafiuddin-Shah M, Tu H-C, Jeffers JR, Zambetti GP, Hsieh JJ-D, Cheng EH. Hierarchical regulation of mitochondrion-dependent apoptosis by BCL-2 subfamilies. Nat. Cell Biol., 8, 1348–1358 (2006).
- 37) Yang E, Zha J, Jockel J, Boise LH, Thompson CB, Korsmeyer SJ. Bad, a heterodimeric partner for Bcl-xL and Bcl-2, displaces bax and promotes cell death. Cell, 80, 285–291 (1995).
- 38) Gross A, McDonnell JM, Korsmeyer SJ. BCL-2 family members and the mitochondria in apoptosis. Genes Dev., 13, 1899–1911 (1999).
- 39) Wolter KG, Hsu Y-T, Smith CL, Nechushtan A, Xi X-G, Youle RJ. Movement of Bax from the cytosol to mitochondria during apoptosis. J. Cell Biol., 139, 1281–1292 (1997).
- 40) Breckenridge DG, Xue D. Regulation of mitochondrial membrane permeabilization by BCL-2 family proteins and caspases. Curr. Opin. Cell Biol., 16, 647–652 (2004).
- 41) Upreti M, Chu R, Galitovskaya E, Smart SK, Chambers TC. Key role for Bak activation and Bak-Bax interaction in the apoptotic response to vinblastine. Mol. Cancer Ther., 7, 2224–2232 (2008).
- 42) Porter AG, Janicke RU. Emerging roles of caspase-3 in apoptosis. Cell Death Differ., 6, 99–104 (1999).
- 43) Li P, Nijhawan D, Wang X. Mitochondrial activation of apoptosis. Cell, 116, S57–S59, 2 p following S59 (2004).
- 44) Walters J, Pop C, Scott FL, Drag M, Swartz P, Mattos C, Salvesen GS, Clark AC. A constitutively active and uninhibitable caspase-3 zymogen efficiently induces apoptosis. Biochem. J., 424, 335–345 (2009).
- 45) Elmore S. Apoptosis: a review of programmed cell death. Toxicol. Pathol., 35, 495–516 (2007).
- 46) Agarwal A, Mahfouz RZ, Sharma RK, Sarkar O, Mangrola D, Mathur PP. Potential biological role of poly(ADP-ribose) polymerase (PARP) in male gametes. Reprod. Biol. Endocrinol., 7, 143–163 (2009).



