MATERIALS AND METHODS
AnimalsMale Wistar rats (Charles-River, Wilmington, MA, U.S.A.) weighing 320 to 380 g were used in the embolic stroke model. Male Sprague-Dawley rats (Harlan, Indianapolis, IN, U.S.A.) weighing 250 to 300 g were used in the ex vivo experiments. All experiments were performed in accord with the NIH Policy and Animal Welfare Act (No. 11/09-179-00) under the approval by Institutional Animal Care and Use Committee (IACUC) at Michigan State University.
MaterialsAA was obtained from Sigma-Aldrich (St. Louis, MO, U.S.A.) and dissolved to a concentration of 50 mg/mL in ubisol-Aqua (Zymes LLC, Hasbrouk Heights, NJ, U.S.A.). Ubisol-Aqua was provided free of charge by Zymes. The lateral tail vein was used for the administration of AA and ubisol-Aqua (vehicle). Recombinant human tPA (Genentech, South San Francisco, CA, U.S.A.) was infused intravenously at a dose of 2.5 mg/kg as a 10% bolus, and the remainder was infused continuously over a 30 min interval with a Harvard pump (Harvard Apparatus, Holliston, MA, U.S.A.) at 2 h after middle cerebral artery occlusion (MCAO).
Preparation of the EmbolusFemoral arterial blood from a donor rat was withdrawn into 20 cm of polyethylene tubing (PE-50) and retained in the tube for 2 h to clot at room temperature and subsequently retained for 22 h at 4°C. Four centimeters of the PE-50 tube containing clot was cut and attached at each end to a 40-mm PE-10 tube interconnected by a syringe filled with saline. The clot was shifted by continuous alternating movement from one syringe to the other for 5 min. A single clot was transferred to a modified PE-50 catheter with a 0.3-mm outer diameter filled with saline.13)
Animal ModelA research investigator blind to the random group assignment performed MCAO surgery. After induction of general anesthesia by isoflurane inhalation, rats were maintained under anesthesia through the surgical period. Rectal temperature was maintained at 37°C with a heating pad. The cerebral blood flow was measured with laser Doppler flowmetry (Perimed, North Royalton, OH, U.S.A.). The MCA was occluded by placement of an embolus at the origin of the MCA. Briefly, under an operating microscope, the right common carotid arteries, right external carotid artery (ECA), and internal carotid artery (ICA) were isolated via a mid line incision. A modified PE-50 catheter with a 0.3-mm outer diameter filled with a single clot, which was attached to a 100 µL Hamilton syringe filled with 0.9% saline, was introduced into the ECA lumen through a small puncture. A 15- to 16-mm length of catheter was gently advanced from the ECA into the lumen of the ICA. The clotin the catheter was injected into the ICA along with 2 to 3 µL of 0.9% saline. The catheter was withdrawn from the right ECA 5 min after injection. The right ECA was ligated.
Neurological ScoreNeurological evaluation was performed by a blinded investigator according to a well-established neurological scoring system consisting of 6 items: spontaneous activity, symmetry of limb movement, forepaw outstretching, climbing, body proprioception and response to vibrissae touch.12,14) Each item was graded from 0 (severe deficit) to 3 (no deficit). The grading of all 6 items was added. The resulting score for each animal can reach a maximum of 18 points in a healthy rat.
Calculation of Infarct Volume Using Triphenyltetrazolium Chloride (TTC) StainingAt 24 h after MCAO surgery, rats were euthanized by isoflurane over dose, decapitated, and the brains were rapidly removed for the determination of infarct size. Brains were cut into 2 mm sections stained with 2% TTC and fixed in 4% formalin. Each section was scanned by a computer scanner to produce digital image, and analyzed using the software program Image/J, available from the NIH. The infarct volume for each section was determined by taking the average of the measured infarct area on each side of the section and multiplying by the section thickness. Edema correction was performed as previously described.12)
Brain Mitochondrial IsolationBrains were rapidly removed at 24 h after intraluminal 6 h tMCAO.8) Whole ipsilateral ischemic and contralateral nonischemic hemispheres were dissected out and placed in homogenizer containing isolation buffer (215 mM mannitol, 75 mM sucrose, 0.1% bovine serum albumin (BSA), 20 mM N-(2-hydroxyethyl)piperazine-N′-2-ethanesulfonic acid (HEPES), 1 mM ethylene glycol bis(2-aminoethylether)-N,N,N′,N′-tetraacetic acid (EGTA), pH adjusted to 7.2 with KOH). The tissue was homogenized, and 30% Percoll in isolation buffer was added. The resultant homogenate was layered on a discontinuous Percoll gradient with the bottom layer containing 40% Percoll solution, followed by a 24% Percoll solution, and finally the sample in a 15% Percoll solution. The density gradients were spun in a Sorvall RC-5C plus superspeed refrigerated centrifuge (Asheville, NC, U.S.A.) in a fixed angle SE-12 rotor at 30400×g for 10 min. Following centrifugation, band 3 (nonsynaptic mitochondria) were separately removed from the density gradient. The final mitochondrial pellet was resuspended in isolation buffer without EGTA to yield a protein concentration of ca. 10 mg protein/mL and stored on ice. Protein concentration was determined using the bicinchoninic acid protein assay (BCA™ Protein Assay Kit, Thermo Scientific, Rockford, IL, U.S.A.).15,16) Total mitochondria from brain tissue were extracted and prepared by using the radio immunoprecipitation assay (RIPA) lysis buffer on ice.
Western Blot AnalysisBriefly, after addition of sample loading buffer, protein samples were electrophoresed on a 15% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and subsequently transferred to nitrocellulose membrane (Millipore, U.S.A.). The membrane was incubated in fresh blocking buffer (0.1% Tween 20 in Tris-buffered saline (TBS), pH 7.4, containing 5% non-fat dried milk) at room temperature for 30 min and then probed with the primary antibody in blocking buffer at 4°C overnight. We used primary antibodies to CytoC (mouse, BD Pharmingen™, 1 : 1000), apoptosis-inducing factor (AIF; rabbit, Epitomics, 1 : 1000), and CytoC oxidase IV (COX IV; rabbit, Cell Signaling, U.S.A., 1 : 1000). The membranewas washed three times for 5 min each using TBST (TBS and 0.1% Tween 20). After that it was incubated in the appropriate horseradish peroxidase (HRP)-conjugated secondary antibody at room temperature for another 1 h and washed again three times in TBST buffer. Membranes were incubated in Super Signal West Pico Chemiluminescent Substrate (Pierce Biotechnology) and exposed to X-ray film. Films were scanned into a computer for measurement of band optical density.
Primary Neuronal CulturesCerebral cortices were isolated from newborn C57BL/6J mice at post-natal day 0.17) Dissociated cells were washed with Neurobasal A and triturated with a pipette, and plated onto poly-D-lysine-precoated plates. Three days after plating, 50% of the medium was changed and subsequently replaced every three days with Neurobasal A supplemented with 2% B27. Neuronal cultures were maintained in a CO2 incubator at 37°C in 5% CO2, and used between DIV 7 and 11. These cultures contained >90% neurons as revealed by NeuN/beta tubulin-immunohistochemistry. For L-glutamate toxicity studies, primary neuronal cells were treated with L-glutamate containing media and incubated at 37°C. To examine AA protection, cells were pretreated with AA for 1 h prior to L-glutamate insults.
Determination of CytotoxicityL-Glutamate-induced cytotoxicity was determined using lactate dehydrogenase (LDH) assay. To detect the loss of membrane integrity, a typical marker of cytotoxicity, the extent of LDH release was measured in conditioned media using the Cytotox 96R Non-Radioactive Cytotoxicity Assay (Promega Corporation). Cell viability in sister cells treated with 50 µM L-glutamate, which induces near complete neuronal death, was used as the total cell death (100%).
Statistical AnalysesAll data were presented as mean±S.E.M. The degree of statistical significance between groups was determined on the basis of ANOVA and post-hoc tests (IBM SPSS statistics 20.2). Post-hoc multiple comparisons used Tukey HSD. A p value of <0.05 value was considered significant.
RESULTS AND DISCUSSION
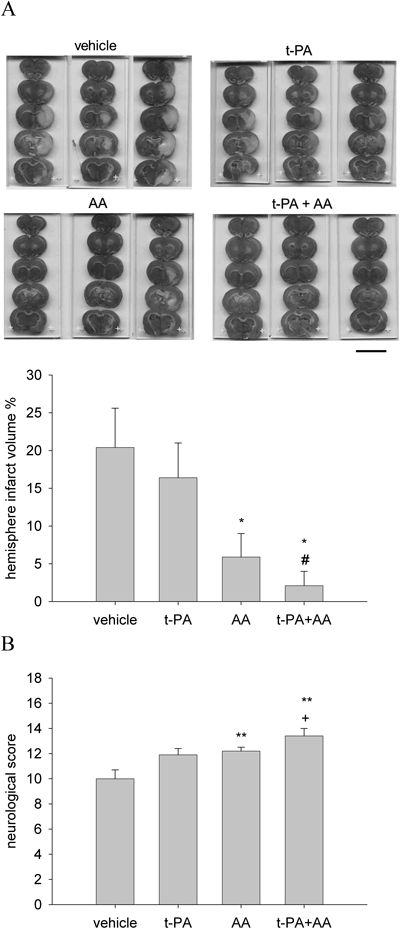
A one-way ANOVA test was performed on infarct volumes [F(3, 37)=4.110, p<0.05] and neurological score [F(3, 37)=6.593, p<0.01]. AA (75 mg/kg, 3 h after MCAO) significantly reduced infarct volume (p=0.025) and neurological deficits (p=0.001) compared to the group treated with vehicle. However, no significant reductions in infarct volume or neurological scores were detected in low-dose t-PA treated rats. Combination treatment with AA and low-dose t-PA resulted in significant reductions of infarct volume (p=0.018) and neurological scores (p=0.004) compared to the group treated with vehicle. Also, combination treatment resulted in further reductions of infarct volume (p=0.014) and neurological scores (p=0.053) compared to the group treated with low-dose t-PA. Combination treatment resulted in significant reductions of only neurological scores (p=0.037) not infarct volume (p=0.387) compared to the group treated with AA (Fig. 1).
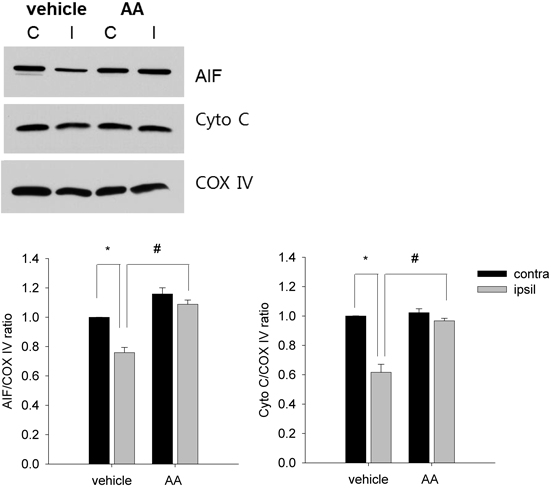
Ischemia-induced mitochondrial dysfunctionis considered to be an important event leading to neuronal apoptosis. To determine the influence of AA on mitochondrial release of CytoC and AIF, Western blot analyses 24 h after ischemia were performed to measure mitochondrial CytoC and AIF. A one-way ANOVA test was performed on CytoC/COX IV ratio [F(3, 7)=36.997, p<0.01] and AIF/COX IV ratio [F(3, 7)=31.571, p<0.01]. Ischemia/reperfusion significantly decreased mitochondrial concentrations of CytoC (p=0.021), and AIF (p=0.020) (Fig. 2). This effect was significantly attenuated by AA treatment: 36% reduction of CytoC release (p=0.019) and 30% reduction of AIF release (p=0.025).
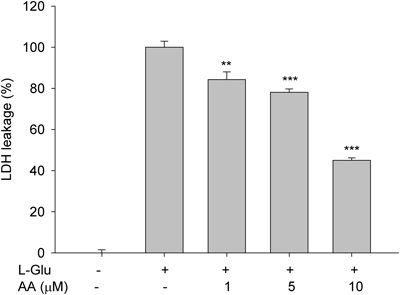
We also determined the effect of AA in primary cortical neurons. L-Glutamate (50 µM) was used to induce neuronal damage. A one-way ANOVA test was performed on LDH leakage [F(4, 35)=251.359, p<0.001]. Pretreatment with AA (1 to 10 µM) was protective against L-glutamate-induced excitotoxicity (Fig. 3).
AA is fast emerging as an exciting candidate for acute stroke therapy. We have previously shown in a mouse model of stroke that AA is neuroprotective, has antioxidant activity and protects blood–brain barrier integrity after ischemia.11) Also, AA is effective against multiple rat models of focal ischemia, has a long therapeutic time window and is effective in females and hypertensive animals. It may mediate neuroprotection by protecting mitochondria and inhibiting matrix metalloproteinase-9 induction and activation.12)
The current study has confirmed efficacy in a rat model of embolic focal cerebral ischemia. Most animal stroke models were developed to induce cerebral ischemia within the MCA territory with the intent of mimicking a common clinical scenario.18) Although the commonly used intraluminal suture MCAO model in the rat has been an indispensable tool for unraveling the complex pathophysiology of permanent and transient focal cerebral ischemia,19) it does not, however, reliably reproduce the inhomogeneous vascular findings at the microvascular and macrovascular level in acute territorial stroke.20) Furthermore, the therapeutic window for therapeutic options may be substantially shorter in this model than in humans.21,22) Because the underlying pathophysiology of brain damage is likewise heterogeneous, embolic MCAO is of increasing interest to study thrombolytic and neuroprotective drugs because it more closely simulates the pathophysiology of human MCAO.23) For these reasons we tested the effect of AA in an embolic stroke model and hypothesized that combining AA with t-PA will significantly enhance neuroprotection. Our results show that combining AA with t-PA enhanced the neuroprotective effect both histologically and functionally.
CytoC and AIF lead to caspase-dependent and caspase-independent apoptotic pathways respectively. In caspase-dependent pathways, released CytoC activates the caspase cascade, causing cleavage of manyproteins, DNA damage, and ultimately cell death.24) Recently it was reported that caspase inhibition abrogated CytoC release in traumatic brain injury.25) Caspase-independent cell death occurs via calpain- or poly(ADP-ribose) polymerase-1-mediated AIF release from the mitochondria, with subsequent translocation to the nucleus.24) Using immunostaining in the mouse brain at 24 h following stroke, we previously reported that AA reduces CytoC release from mitochondria. Our previous in vitro data also showed that AA was indeed capable of markedly reducing CytoC release from isolated brain mitochondria preparations exposed to elevated calcium levels, H2O2, or nitric oxide.11) The present results show that AA reduced CytoC, and AIF release from mitochondria thus confirming our previous findings. These data support the pleitropic effects of AA against brain ischemia.
In conclusion, our data suggest that treatment with AA at 3 h exerts a neuroprotective effect after embolic stroke. The combination of AA with the low-dose tPA markedly improved the neuroprotective effect, which represents a promising approach for the treatment of stroke.