Abstract
The effects of paroxetine, a selective serotonin reuptake inhibitor, on human ether-a-go-go-related gene (HERG) channels were investigated using the whole-cell patch-clamp technique. The HERG channels were stably expressed in human embryonic kidney cells. Paroxetine inhibited the peak tail currents of the HERG channel in a concentration-dependent manner, with an IC50 value of 0.45 µM and a Hill coefficient of 0.85. These effects were reversible after wash-out of the drug. The paroxetine-induced inhibition of the HERG channels was voltage-dependent. There was a steep increase in inhibition over the voltage range of the channel opening. Also, a shallow voltage-dependent inhibition was detected over the voltage range in which the channels were fully activated. The fractional electrical distance was estimated to be 0.11. Paroxetine induced a leftward shift in the voltage-dependence of the steady-state activation of the HERG channels. Before and after application of the 1 µM paroxetine, the half-maximum activation was −14.21 mV and −27.04 mV, respectively, with no shift in the slope value. The HERG channel block was not use-dependent. The characteristics of the block were dependent on open and inactivated channel states rather than closed state. Paroxetine had no effect on activation and deactivation kinetics, steady-state inactivation. These results suggest that paroxetine blocks the HERG channels by binding to these channels in the open and inactivated states.
Treating depression with selective serotonin reuptake inhibitors (SSRIs) is thought to be safer than using tricyclic antidepressants (TCAs) with regard to cardiovascular side effects. Since TCAs are known to induce electrocardiographic QT prolongation, their use also has an associated arrhythmogenic risk in vivo.1–4) Paroxetine, one of the SSRIs (Fig. 1), has the most potent activity in the inhibition of the 5-hydroxytryptamine (5-HT) uptake transporter.5,6) The efficacy of paroxetine for depression is comparable to that of TCAs. Since there are fewer side effects and the lower toxicity with paroxetine,7,8) it has become an attractive drug for the effective treatment of depressive patients with cardiovascular problems. However, accumulating evidence indicates that paroxetine has various additional effects, especially on several ion channels, in a SSRI-independent manner. For example, paroxetine has been shown to act as a potent Na+ channel blocker.9–11) It has also been reported that paroxetine inhibits the G protein-activated inwardly rectifying K+ channel12) and two P domain background K+ channel, TREK.13) These results suggest that paroxetine may have an effect on the cardiovascular system. However, the side effects of paroxetine on the cardiovascular system have not been thoroughly studied.
In ventricular myocytes, reduced function of the cardiac K+ current (IK) can cause long QT syndrome. The IK includes the rapidly (IKr) and slowly (IKs) activating components of the delayed rectifier K+ current.14,15) The IKr is encoded by the human ether-a-go-go-related gene (HERG). Mutation in the HERG has been shown to cause chromosome 7-linked inherited long QT syndrome (LQT2), which is related to Torsades de pointes, a ventricular tachyarrhythmia that can potentially lead to sudden death.16,17) Several drugs that block the HERG channels have been shown to cause cardiotoxicity.18–20)
In this study, we investigated the effects of paroxetine on the HERG channels stably expressed in human embryonic kidney (HEK293) cells. We also characterized the nature of the action on HERG channels by paroxetine.
MATERIALS AND METHODS
Cell Culture and TransfectionHEK293 cells stably expressing HERG channels, a kind gift from January,21) were used for electrophysiological recordings. The method for establishing the HERG channels expression in HEK293 cells is briefly described as follows. HERG cDNA was transferred into the plasmid expression vector pCDNA3 vector (Invitrogen Corporation, San Diego, CA, U.S.A.). HEK293 cells were stably transfected with HERG cDNA using a calcium phosphate precipitate method (Invitrogen) or a lipofectamine method (Invitrogen). The transfected cells were cultured in minimum essential medium (MEM) supplemented with 10% fetal bovine serum, 1 mM sodium pyruvate, 0.1 mM non-essential amino-acid solution, 100 units/mL penicillin, and 100 µg/mL streptomycin sulfate. The cultures were passaged every 4–5 d with a brief trypsin–ethylenediaminetetraacetic acid (EDTA) treatment followed by seeding onto glass coverslips (diameter: 12 mm, Fisher Scientific, Pittsburgh, PA, U.S.A.) in a Petri dish. After 12–24 h, the cell-attached coverslips were used for electrophysiological recordings.
ElectrophysiologyThe HERG currents were recorded from HEK293 cells, with a whole-cell patch-clamp technique22) at room temperature (22–23°C). The micropipettes fabricated from glass capillary tubing (PG10165-4; World Precision Instruments, Sarasota, FL, U.S.A.) with a double-stage vertical puller (PC-10; Narishige, Tokyo, Japan) had a tip resistance of 2–3 MOhm, when filled with the pipette solution. The whole-cell currents were amplified with Axopatch 1D amplifier (Molecular Devices, Sunnyvale, CA, U.S.A.), digitized with Digidata 1200A (Molecular Devices) at 5 kHz and low-pass filtered with four-pole Bessel filter at 2 kHz. The capacitive currents were canceled and series resistance was compensated at 80% with the amplifier, while leak subtraction was not used. The generation of voltage commands and acquisition of data were controlled with pClamp 10.03 software (Molecular Devices) running on an IBM-compatible Pentium computer. The recording chamber (RC-13, Warner Instrument Corporation, Hamden, CT, U.S.A.) was continuously perfused with bath solution (see below for composition) at a rate of 1 mL/min.
Solutions and DrugsThe external solution contained 137 mM NaCl, 4 mM KCl, 1.8 mM CaCl2 1 mM MgCl2, 10 mM glucose, and 10 mM N-(2-hydroxyethyl)piperazine-N′-2-ethanesulfonic acid (HEPES) (adjusted to pH 7.4 with NaOH). The intracellular solution contained 130 mM KCl, 1 mM MgCl2, 5 mM ethylene glycol bis(2-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA), 5 mM MgATP, and 10 mM HEPES (adjusted to pH 7.4 with KOH). Paroxetine (Sigma Chemical Co., St. Louis, MO, U.S.A.) was dissolved in ethanol at 30 mM and further diluted into the bath solution. The final bath solution concentration of ethanol (less than 0.1%) had no effect on HERG currents (data not shown).
Data AnalysisData were analyzed with Origin 7.0 (OriginLab Corp., Northampton, MA, U.S.A.) and Clampfit 10.03 software (Molecular Devices). An IC50 and Hill coefficient (n) were obtained by fitting concentration dependence data to the following equation:
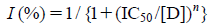 | (1) |
I (%) is the percent inhibition of current (I (%)=[1−Idrug/Icontrol]×100) at test potential, and [D] represents various drug concentrations. The steady-state activation/inactivation curves were fitted with the Boltzmann equation:
 | (2) |
k represents the slope factor, V the test potential, and V1/2 the potential at which the conductance was half-maximal. Deactivation was fitted to a biexponential process, as follows:
 | (3) |
in which τ1 and τ2 are the time constants; A1 and A2 are the amplitudes of each component of the exponential; and B is the baseline value.
To investigate the voltage dependence of HERG inhibition by the drug, the relative current was plotted as a function of the membrane potential. The resultant percent inhibition data between +10 and +60 mV were fitted with a Woodhull equation23):
 | (4) |
KD(0) represents the apparent affinity at 0 mV, z the charge valence of the drug, δ the fractional electrical distance, F Faraday’s constant, R the gas constant, and T the absolute temperature. A value of 25.4 mV was used for RT/F at 23°C in the present study.
Results were expressed as mean±S.E.M. Student’s t-test and ANOVA were used for statistical analysis. A value of p<0.05 was considered statistically significant.
Virtual DockingThe three-dimensional structure of paroxetine was built using LigPrep (Schrödinger, LLC, New York, NY, U.S.A.). The virtual docking of paroxetine to the HERG channel was performed by Glide (Schrödinger). The PoseView (BioSolveIT GmbH, Sankt Augustin, Germany) was used to investigate the interactions of the paroxetine molecule with the HERG channel. All structural figures were prepared using PyMOL v1.2 (DeLano Scientific LLC; San Francisco, CA, U.S.A.) and ChemDraw Ultra 11.0 (CambridgeSoft; Cambridge, MA, U.S.A.).
RESULTS
Inhibition of HERG-Mediated Current (IHERG) by ParoxetineWe examined the effects of paroxetine on IHERG, using a whole-cell patch-clamp technique. As shown in Fig. 1A (upper trace), the whole-cell currents were elicited with a 4-s depolarization to +20 mV, from a holding potential of −80 mV. The tail current was recorded at −50 mV for 6 s in HEK293 cells expressing HERG channels. The any currents including the whole-cell and tail currents was not detected in non-transfected HEK293 cells using the same protocols (data not shown). Bath-applied paroxetine reduced the IHERG in a concentration-dependent manner (Fig. 1A) and steady-state inhibition was reached within 3 min. As shown in Fig. 1B, dose dependence of the steady-state currents measured at the end-pulse of +20 mV or peak tail currents was analyzed quantitatively. A nonlinear least-squares fit of dose–response plots with the Hill equation yielded an IC50 value of 0.35±0.01 µM and a Hill coefficient of 0.80±0.01 (n=5) for the steady-state currents and an IC50 value of 0.45±0.01 µM and a Hill coefficient of 0.85±0.01 (n=5) for the peak tail currents. Figure 1C shows the effects of 1 µM paroxetine on the tail peak currents during a drug wash-in and wash-out process. The tail current was obtained by applying a 4-s depolarizing pulse to +20 mV from a holding potential of −80 mV, followed by a repolarizing pulse to −50 mV for 6 s every 15 s. The inhibition of the tail peak current by paroxetine occurred rapidly to reach a steady-state level within 3 min. The inhibition was reversible as the current amplitudes rose to the baseline values upon wash-out of the drug (Fig. 1C). The tail peak currents that were reduced by 1 µM paroxetine were reversed to 83.21±4.11% of pre-drug baseline (n=5) after 3-min wash-out.
Voltage Dependence of Paroxetine-Mediated Inhibition of IHERGWe next investigated the IHERG–voltage (I–V) relationship (Fig. 2). IHERG was produced by applying 4-s depolarizing pulses between −70 and +60 mV in 10-mV increments every 15 s from a holding potential of −80 mV. The tail current was recorded at −50 mV for 6 s. Figure 2A shows representative superimposed current traces recorded under control conditions and 3 min after exposure to 1 µM paroxetine in the same cell. The normalized I–V relationships for IHERG measured at the end of the depolarizing pulses and for peak tail current are shown in Figs. 2B and C, respectively. Under control conditions, the IHERG measured at the end of the depolarizing pulses began to be generated at −40 mV and peaked at about 0±10 mV and decreased thereafter, resulting in a negative slope of the I–V curve, while the tail currents are fully activated following pulses to 10±60 mV. In the presence of 1 µM paroxetine, IHERG measured at the end of the depolarizing pulses and tail current amplitudes were reduced through the whole voltages ranges which HERG channel was activated. But, at −40 and −30 mV, slight increases of IHERG by the drug were detected. To examine the effect of paroxetine on voltage-dependent activation of HERG channels, the tail peak currents were normalized and plotted against the membrane potential (Fig. 2C). Data were fitted with a Boltzmann function. The voltage of half-maximum activation (V1/2) was −14.21±1.34 mV (n=5) and −27.04±2.71 mV (n=5) before and after application of the 1 µM paroxetine showing that the V1/2 was significantly shifted leftward. However, there was no shift in slope value (k) in the presence of 1 µM paroxetine (6.94±0.18 mV for control; 7.30±0.34 mV for paroxetine, n=5).
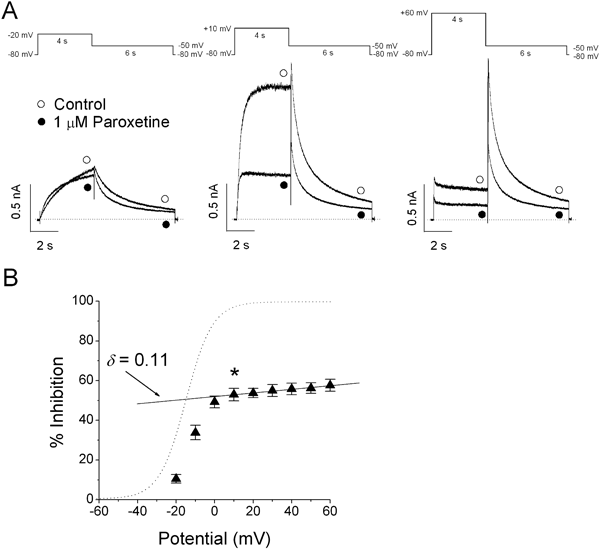
To examine the voltage dependence of the paroxetine effect, we compared the extent of percent inhibition of IHERG (see Materials and Methods) at different potentials. Figure 3A shows representative superimposed current traces under control conditions and in the presence 1 µM paroxetine selected at three different potentials. Figure 3B shows the relative % inhibition of IHERG against membrane potential. A high degree of inhibition with a strong voltage dependence was observed between −20 and +10 mV. This voltage range is the voltage range of HERG channel opening (Fig. 3B). Between +10 and +60 mV, despite the HERG channel being fully activated at this voltage range, inhibition continued to increase with a shallow voltage dependence: 52.91±3.21% inhibition at +10 mV, and 57.71±3.02% inhibition at +60 mV (n=5, p<0.05). Paroxetine is positively charged at an intracellular pH of 7.3 (pKa=10.3). Given the assumption that paroxetine interacts intracellularly with HERG channel, the further increase of inhibition over the voltage range where the channels are fully activated might be explained by the induction of heightened outward driving forces at the binding site, due to increasing positive intracellular potentials. We investigated this effect by a linear curve fitting of the data at potentials positive to +10 mV, using a linear transformation of the Woodhull equation (see Materials and Methods). The solid line in Fig. 3B represents a fitted curve which yielded a fractional electrical distance of δ=0.11±0.01 (n=5).
State Dependence of Paroxetine-Mediated Inhibition of IHERGTo examine the state dependence (closed/activated or open/inactivated) of channel block, we activated currents under control conditions and in the presence of the drug using a single depolarizing step to 0 mV for 8 s (Fig. 4A). Having obtaining a control measurement, 1 µM paroxetine was applied to the cell for 3 min to allow an equilibration of drug concentrations within the bath and the cell. For this experiment, cells were held continuously at −80 mV to the channels in the closed state. Then the depolarization protocols resumed. Figure 4B shows the time course of inhibition by plotting percent inhibition of IHERG (see Materials and Methods) against time. The time-dependent development of IHERG inhibition by paroxetine was detected. The time-dependent inhibition at the end of 0 mV pulse increased up to 54.41±7.21% (n=4). This result suggests that IHERG is little blocked by paroxetine when the HERG channels are predominantly in the closed state. Therefore, the block of IHERG by paroxetine requires channel activation from the closed state.
In order to examine whether the HERG channels in their inactivated state are blocked by paroxetine, a long test pulse to +80 mV for 4 s was applied to inactivate the channels. This long test pulse was followed by a second voltage step of 0 mV for 4 s to open the HERG channels. Figure 4C shows representative current traces under control conditions and after a 3-min treatment with 1 µM paroxetine, while holding the cell at −80 mV. As shown in Fig. 4D, the normalized fractional inhibition upon channel opening during the second voltage step (0 mV) is not time-dependent, indicating no additional inhibition, while the channels were re-activated. The inhibition at the end of second pulse (0 mV) was 56.61±5.81% (n=4). This result suggests that the pronounced inhibition of the IHERG had already been reached during the previous inactivating 80-mV pulse. On the basis of the above overall data (Fig. 4), paroxetine blocks the HERG channels predominantly in the open and inactivated state, rather than in the closed state.
Frequency-Independent Inhibition of IHERG by ParoxetineTo examine the frequency dependence of IHERG inhibition by paroxetine, IHERG was elicited by a 300-ms depolarization to +20 mV from a holding potential of −80 mV, followed by a 300-ms repolarization to −50 mV, 15 times at 0.2 and 1 Hz. The frequency-dependent inhibition of the steady-state currents measured at the end-pulse of +20 mV was analyzed by normalization with the first current amplitudes obtained at the first pulse. Figure 5A shows representative current traces under control conditions and after a 3 min application of 1 µM paroxetine. As shown in Figs. 5A and B, under control conditions and in the presence of 1 µM paroxetine, the relative IHERG was reduced only to a slight extent at 0.2 and 1 Hz stimuli. This result suggests that neither the development of inhibition nor the amount of steady-state inhibition was frequency-dependent (n=5).
Effects of Paroxetine on the Kinetics of Channel GatingWe examined whether paroxetine has an effect on activation, deactivation kinetics and steady-state inactivation of HERG channel. To test the time course of HERG channel activation, IHERG was obtained by a depolarizing pulse of +20 mV from a holding potential of −80 mV for varying durations, followed by a 1-s repolarizing pulse of −50 mV. To evaluate the HERG channel activation, the time constant was obtained by a single-exponential fitting to the envelope of tail currents (Figs. 6A, B). The time constant for activation was not affected by paroxetine: 209.22±21.21 ms under control conditions and 197.21±17.31 ms in the presence of 1 µM paroxetine, respectively (n=5). Deactivation current was obtained by 5-s repolarizing pulses from −90 to −30 mV, increments of 10 mV after a 1 s depolarizing pulse of +60 mV from a holding potential of −80 mV (Figs. 6C, D). Deactivation time constants were obtained by a double-exponential fitting to the decay traces of tail currents. The two time constants, fast and slow components, were not affected by paroxetine. Steady-state inactivation was examined using a double-pulse protocol with varying interstimulus repolarizing pulses (Figs. 6E, F). After the first 200-ms depolarizing pulse of +60 mV induced HERG channel inactivation, 10-ms interstimulus pulses from −100 to +40 mV with a 10 mV increment were applied, followed by a second depolarizing pulse of +60 mV. Peak current amplitudes were measured during the second depolarizing pulse of +60 mV and normalized data by the maximum values were plotted as a function of the interstimulus pulses. By fitting data to the Boltzmann function, the inactivation curves were not changed by paroxetine. The potential of half-maximum activation (V1/2) was –12.08±1.23 mV and −13.81±1.11 mV before and after application of 1 µM paroxetine (n=5). Similarly, there was no shift in slope value (k) in the presence of the drug (30.42±1.11 mV for control; 31.32±1.21 mV for paroxetine, n=5). In summary, paroxetine did not show any effect on activation, deactivation kinetics and steady-state inactivation.
Docking of Paroxetine within the Inner Cavity of a HERG Channel Homology ModelWe have two hypotheses about the binding mode of HERG channel blockers. The first hypothesis holds that protonated nitrogen of the HERG channel inhibitors would form a cation–π interaction with the aromatic ring of HERG residue Y652 and hydrophobic interaction with HERG residue F656. However, this hypothesis does not account for reports that residue T623 is also critical to high-affinity HERG binding.24) We performed virtual docking of paroxetine in the binding pocket of the HERG channel, using a Glide program. Our docking result is shown in Fig. 7. A detailed analysis of the receptor–ligand interaction using the PoseView program showed that the hydrophobic contacts were made between paroxetine and Y652. The protonated nitrogen of paroxetine makes a hydrogen bond with the carbonyl oxygen of residue T623D. Based on this data, we propose a second hypothesis. The protonated nitrogen of a blocker makes a hydrogen bond with the carbonyl oxygen of residue T623, an aromatic moiety of the blocker makes a π–π interaction with the aromatic ring of residue Y652. Finally, a hydrophobic group of the blocker makes a hydrophobic interaction with the benzene ring of residue F656.25)
DISCUSSION
We show that paroxetine blocks the HERG channels expressed in HEK293 cells, as measured using a patch-clamp technique.
The paroxetine-induced blockage is mixed state-dependent. The paroxetine-induced inhibition of the HERG channel was associated with a time-dependent development of the blockade. When the HERG channels are predominantly in the closed state, the IHERG is little blocked by paroxetine. The block of IHERG by paroxetine requires channel activation from the closed state. The open-state dependent inhibition of the HERG channels by paroxetine is supported by the result that the inhibition of IHERG by paroxetine is highly voltage-dependent and increased steeply in the voltage range of channel activation. The voltage-dependent inhibition of IHERG by paroxetine supports the model that IHERG inhibition by paroxetine preferentially occurs after channels are open. Additionally, the pronounced inhibition of the IHERG by paroxetine that is reached during the inactivating process, suggests that paroxetine inhibits the HERG channels predominantly in the inactivated state. These results suggest that paroxetine blocks the HERG channels predominantly in the open and inactivated states rather than in the closed state.
However, we cannot completely rule out the possibility of the closed-state channel block. A five-state model has been suggested for HERG channels, including an open, an inactivated and several closed states.26) Although many drugs have been tested so far, the inhibition was shown to occur in the open and inactivated, but not in the closed states.27–29) In the present study, the inhibition of the IHERG by paroxetine was not frequency-dependent. Although the much higher association and dissociation rate constant of paroxetine presumably contributed to the lack of frequency-dependent inhibition of HERG channels in open-state blocking mechanism, it is usually detected in open-state channel blockade. Furthermore, the lack of IHERG inhibition was detected at the voltage ranges between −40 and −20 mV, resulting in the leftward shift in voltage dependence of steady-state activation. This result indicates more current would be significantly detected on depolarization than that in the absence of paroxetine. This net increase in current could conceivably mask the effects of closed-channel blockade at these voltages. Therefore, it is difficult to conclude that paroxetine interacts with pure open- or inactivated state of HERG channel. The voltage-dependent inhibition of IHERG by antiarrhythmic drug, amiodarone and antiparkinsonian drug, budipine with mixed-state dependence on closed-, open- and inactivated states were reported previously.30,31 Fluvoxamine and azimilide have been reported to exert an agonist action at negative voltages, while inhibiting the current at more positive potentials.31–33) Those drugs also induced a leftward shift in voltage dependence of steady-state activation. These results are similar to our data. Furthermore, the result of virtual docking suggests that a hydrophobic group of the blocker makes a hydrophobic interaction with the benzene ring of residue F656.25) This kind of the hydrophobic interaction may affect the gating kinetics of the transition from the closed to the open states, which indicates the interaction between blocker and closed-state HERG channel. Therefore, on the basis of previous reports and the results of this study, paroxetine may interact with the closed-state of the HERG channel in this study.
It has been reported that two amino acids, Y652 and F656, are important for the blocking of the HERG channel using an alanine-scanning mutagenesis study34) and that a protonated nitrogen of HERG channel blockers may form a cation–π interaction with the aromatic ring of HERG residue Y652 and a hydrophobic interaction with HERG residue F656.24) In the present study, the docking simulation results are consistent with our previous HERG channel block model that the protonated nitrogen of a blocker makes a hydrogen bond with the carbonyl oxygen of residue T623 or S624, that an aromatic moiety of the blocker makes a π–π interaction with the aromatic ring of residue Y652, and that a hydrophobic group of the blocker makes a hydrophobic interaction with the benzene ring of residue F656.25) However, due to the limitation of the current computer simulation methods, the virtual docking result has limitations. Because there is no known HERG channel structure, a homology model of HERG channel was used, which may result in inaccurate docking. Also an empirical force field was used, which would approximate the docking process. Therefore, to confirm the molecular interaction suggested in this study, it would be necessary to perform a test using mutant HERG channels like F656A and Y652A.
On the basis of the pharmacokinetics of paroxetine, the therapeutic plasma concentrations are reported to be 0.06–0.18 µM in depressed patients.35) In the present study, the IC50 value (0.35–0.45 µM) of paroxetine for blocking IHERG is higher than the therapeutic plasma concentrations. However, significant inhibition of IHERG at concentrations of 0.1 µM paroxetine was detected. Furthermore, the effects of paroxetine on the HERG channel were examined in a HEK293 cell line. The phospholipid composition of the cell may be different from that of human native cardiac myocytes, and the differences in membrane composition may affect paroxetine-induced IHERG blockade. Also, drug concentrations in tissues may be higher than in plasma due to paroxetine’s high lipophilicity and affinity for adipose tissues.36,37) Furthermore, there are several reports showing that the expression level of HERG channel is reduced by long time exposure of SSRIs.38,39) Drugs inducing HERG channel blockade are likely to be identified by standard electrophysiological method, for example, patch-clamp techniques. But, trafficking effects may be missed. Thus, standard electrophysiological method alone could be lead to an underestimation of the net effect on HERG channel by a drug. Therefore, it is possible that in this study, the extent of the blocking effects of paroxetine on IHERG under physiological conditions may be underestimated. The paroxetine-induced block of IHERG could be clinically relevant in the upper range of therapeutic plasma concentrations that are observed in paroxetine treatment for depressed patients. Therefore, the HERG channel in the heart might be blocked by paroxetine at clinically relevant plasma concentrations, which can contribute to its proarrhythmic effects, such as QT prolongation and polymorphous ventricular tachycardia.16,17,40,41)
In conclusion, the present study has described, for the first time, the effects of paroxetine on the HERG channel expressed in HEK293 cells. Detailed study of the interaction kinetics between paroxetine and HERG channel suggests that the blockade of the HERG channel by paroxetine is dependent on open and inactivated channel states and possibly event interaction with the closed state of the HERG channel. Thus, much caution about cardiotoxicity is required, when using paroxetine in the treatment of depressed patients who have cardiovascular disease as a comorbidity.
Acknowledgment
The authors wish to thank Dr. Craig January (Section of Cardiology, University of Wisconsin, Madison, WI, U.S.A.) for providing the HERG-expressing HEK293 stable cell line. This research was supported by a grant (08172KFDA465) from Korea Food & Drug Administration in 2008 and by a grant (09172KFDA670) from Korea Food & Drug Administration in 2009, and by 2013 Research Grant from Kangwon National University (No. 120131437).
REFERENCES
- 1) Henry JA, Alexander CA, Sener EK. Relative mortality from overdose of antidepressants. BMJ, 310, 221–224 (1995).
- 2) Henry JA. Epidemiology and relative toxicity of antidepressant drugs in overdose. Drug Saf., 16, 374–390 (1997).
- 3) Reilly JG, Ayis SA, Ferrier IN, Jones SJ, Thomas SH. QTc-interval abnormalities and psychotropic drug therapy in psychiatric patients. Lancet, 355, 1048–1052 (2000).
- 4) Scherer D, von Löwenstern K, Zitron E, Scholz EP, Bloehs R, Kathöfer S, Thomas D, Bauer A, Katus HA, Karle CA, Kiesecker C. Inhibition of cardiac hERG potassium channels by tetracyclic antidepressant mianserin. Naunyn Schmiedebergs Arch. Pharmacol., 378, 73–83 (2008).
- 5) Hyttel J. Citalopram—pharmacological profile of a specific serotonin uptake inhibitor with antidepressant activity. Prog. Neuropsychopharmacol. Biol. Psychiatry, 6, 277–295 (1982).
- 6) Wong DT, Bymaster FP, Engleman EA. Prozac (fluoxetine, Lilly 110140), the first selective serotonin uptake inhibitor and an antidepressant drug: twenty years since its first publication. Life Sci., 57, 411–441 (1995).
- 7) Anderson IM. Selective serotonin reuptake inhibitors versus tricyclic antidepressants: a meta-analysis of efficacy and tolerability. J. Affect. Disord., 58, 19–36 (2000).
- 8) Montgomery SA. A meta-analysis of the efficacy and tolerability of paroxetine versus tricyclic antidepressants in the treatment of major depression. Int. Clin. Psychopharmacol., 16, 169–178 (2001).
- 9) Wang GK, Mitchell J, Wang SY. Block of persistent late Na+ currents by antidepressant sertraline and paroxetine. J. Membr. Biol., 222, 79–90 (2008).
- 10) Dick IE, Brochu RM, Purohit Y, Kaczorowski GJ, Martin WJ, Priest BT. Sodium channel blockade may contribute to the analgesic efficacy of antidepressants. J. Pain, 8, 315–324 (2007).
- 11) Huang CJ, Harootunian A, Maher MP, Quan C, Raj CD, McCormack K, Numann R, Negulescu PA, Gonzalez JE. Characterization of voltage-gated sodium-channel blockers by electrical stimulation and fluorescence detection of membrane potential. Nat. Biotechnol., 24, 439–446 (2006).
- 12) Kobayashi T, Washiyama K, Ikeda K. Inhibition of G protein-activated inwardly rectifying K+ channels by the antidepressant paroxetine. J. Pharmacol. Sci., 102, 278–287 (2006).
- 13) Thümmler S, Duprat F, Lazdunski M. Antipsychotics inhibit TREK but not TRAAK channels. Biochem. Biophys. Res. Commun., 354, 284–289 (2007).
- 14) Sanguinetti MC, Jurkiewicz NK. Two components of cardiac delayed rectifier K+ current. Differential sensitivity to block by class III antiarrhythmic agents. J. Gen. Physiol., 96, 195–215 (1990).
- 15) Trudeau MC, Warmke JW, Ganetzky B, Robertson GA. HERG, a human inward rectifier in the voltage-gated potassium channel family. Science, 269, 92–95 (1995).
- 16) Curran ME, Splawski I, Timothy KW, Vincent GM, Green ED, Keating MT. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell, 80, 795–803 (1995).
- 17) Viskin S. Long QT syndromes and torsade de pointes. Lancet, 354, 1625–1633 (1999).
- 18) Suessbrich H, Waldegger S, Lang F, Busch AE. Blockade of HERG channels expressed in Xenopus oocytes by the histamine receptor antagonists terfenadine and astemizole. FEBS Lett., 385, 77–80 (1996).
- 19) Taglialatela M, Castaldo P, Pannaccione A, Giorgio G, Annunziato L. Human ether-a-go-go-related gene (HERG) K+ channels as pharmacological targets: present and future implications. Biochem. Pharmacol., 55, 1741–1746 (1998).
- 20) Suessbrich H, Schonherr R, Heinemann SH, Attali B, Lang F, Busch AE. The inhibitory effect of the antipsychotic drug haloperidol on HERG potassium channels expressed in Xenopus oocytes. Br. J. Pharmacol., 120, 968–974 (1997).
- 21) Zhou Z, Gong Q, Ye B, Fan Z, Makielski JC, Robertson GA, January CT. Properties of HERG channels stably expressed in HEK 293 cells studied at physiological temperature. Biophys. J., 74, 230–241 (1998).
- 22) Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch., 391, 85–100 (1981).
- 23) Woodhull AM. Ionic blockage of sodium channels in nerve. J. Gen. Physiol., 61, 687–708 (1973).
- 24) Sanguinetti MC, Chen J, Fernandez D, Kamiya K, Mitcheson J, Sanchez-Chapula JA. Physicochemical basis for binding and voltage-dependent block of hERG channels by structurally diverse drugs. Novartis Found. Symp., 266, 159–166, discussion, 166–170 (2005).
- 25) Choe H, Nah KH, Lee SN, Lee HS, Lee HS, Jo SH, Leem CH, Jang YJ. A novel hypothesis for the binding mode of HERG channel blockers. Biochem. Biophys. Res. Commun., 344, 72–78 (2006).
- 26) Kiehn J, Lacerda AE, Brown AM. Pathways of HERG inactivation. Am. J. Physiol., 277, H199–H210 (1999).
- 27) Scholz EP, Zitron E, Kiesecker C, Lück S, Thomas D, Kathöfer S, Kreye VA, Katus HA, Kiehn J, Schoels W, Karle CA. Inhibition of cardiac HERG channels by grapefruit flavonoid naringenin: implications for the influence of dietary compounds on cardiac repolarisation. Naunyn Schmiedebergs Arch. Pharmacol., 371, 516–525 (2005).
- 28) Scholz EP, Zitron E, Kiesecker C, Lueck S, Kathöfer S, Thomas D, Weretka S, Peth S, Kreye VAW, Schoels W, Katus HA, Kiehn J, Karle CA. Drug binding to aromatic residues in the HERG channel pore cavity as possible explanation for acquired long QT syndrome by antiparkinsonian drug budipine. Naunyn Schmiedebergs Arch. Pharmacol., 368, 404–414 (2003).
- 29) Zitron E, Karle CA, Wendt-Nordahl G, Kathöfer S, Zhang W, Thomas D, Weretka S, Kiehn J. Bertosamil blocks HERG potassium channels in their open and inactivated states. Br. J. Pharmacol., 137, 221–228 (2002).
- 30) Kiehn J, Thomas D, Karle CA, Schöls W, Kübler W. Inhibitory effects of the class III antiarrhythmic drug amiodarone on cloned HERG potassium channels. Naunyn Schmiedebergs Arch. Pharmacol., 359, 212–219 (1999).
- 31) Milnes JT, Crociani O, Arcangeli A, Hancox JC, Witchel HJ. Blockade of HERG potassium currents by fluvoxamine: incomplete attenuation by S6 mutations at F656 or Y652. Br. J. Pharmacol., 139, 887–898 (2003).
- 32) Jiang M, Dun W, Fan JS, Tseng GN. Use-dependent ‘agonist’ effect of azimilide on the HERG channel. J. Pharmacol. Exp. Ther., 291, 1324–1336 (1999).
- 33) Walker BD, Singleton CB, Tie H, Bursill JA, Wyse KR, Valenzuela SM, Breit SN, Campbell TJ. Comparative effects of azimilide and ambasilide on the human ether-a-go-go-related gene (HERG) potassium channel. Cardiovasc. Res., 48, 44–58 (2000).
- 34) Mitcheson JS, Chen J, Lin M, Culberson C, Sanguinetti MC. A structural basis for drug-induced long QT syndrome. Proc. Natl. Acad. Sci. U.S.A., 97, 12329–12333 (2000).
- 35) Tomita T, Yasui-Furukori N, Nakagami T, Tsuchimine S, Ishioka M, Kaneda A, Nakamura K, Kaneko S. Therapeutic reference range for plasma concentrations of paroxetine in patients with major depressive disorders. Ther. Drug Monit., 36, 480–485 (2014).
- 36) Neugebauer G, Akpan W, von Möllendorff E, Neubert P, Reiff K. Pharmacokinetics and disposition of carvedilol in humans. J. Cardiovasc. Pharmacol., 10 (Suppl. 11), S85–S88 (1987).
- 37) Tomlinson B, Prichard BN, Graham BR, Walden RJ. Clinical pharmacology of carvedilol. Clin. Investig., 70 (Suppl. 1), S27–S36 (1992).
- 38) Hancox JC, Mitcheson JS. Combined hERG channel inhibition and disruption of trafficking in drug-induced long QT syndrome by fluoxetine: a case-study in cardiac safety pharmacology. Br. J. Pharmacol., 149, 457–459 (2006).
- 39) Rajamani S, Eckhardt LL, Valdivia CR, Klemens CA, Gillman BM, Anderson CL, Holzem KM, Delisle BP, Anson BD, Makielski JC, January CT. Drug-induced long QT syndrome: hERG K+ channel block and disruption of protein trafficking by fluoxetine and norfluoxetine. Br. J. Pharmacol., 149, 481–489 (2006).
- 40) Flugelman MY, Pollack S, Hammerman H, Riss E, Barzilai D. Congenital prolongation of Q-T interval: a family study of three generations. Cardiology, 69, 170–174 (1982).
- 41) Rodriguez de la Torre B, Dreher J, Malevany I, Bagli M, Kolbinger M, Omran H, Lüderitz B, Rao ML. Serum levels and cardiovascular effects of tricyclic antidepressants and selective serotonin reuptake inhibitors in depressed patients. Ther. Drug Monit., 23, 435–440 (2001).