Regular Articles
Losartan Competitively Inhibits CYP2C8-Dependent Paclitaxel Metabolism in Vitro
2014 年 37 巻 9 号 p. 1550-1554
詳細
2014 年 37 巻 9 号 p. 1550-1554
The present study aimed to characterize the inhibitory effects of losartan, an angiotensin II receptor blocker, on CYP2C8. Inhibition experiments were based on human lymphoblast-expressed recombinant CYP2C8 (rCYP2C8) and paclitaxel as a CYP2C8 substrate. The disappearance of paclitaxel (initial concentration: 7.5 µmol/L) was monitored over time at different concentrations of losartan (0, 100, 500 and 1000 µmol/L). For Dixon and Cornish–Bowden plots, various concentrations of losartan (final concentration: 0, 50, 100 and 250 µmol/L) and paclitaxel (final concentration: 3.75, 7.5 and 15 µmol/L) were used. Losartan exhibited significant inhibitory effects on paclitaxel disappearance at losartan concentrations of ≥100 µmol/L (p<0.05). Losartan at 50 µmol/L inhibited the disappearance of paclitaxel by about 60%. Both plots showed that losartan exerted competitive inhibition of rCYP2C8, and its apparent Ki value was estimated to be 40.7 µmol/L. The degree of inhibition (R value) for rCYP2C8 after oral administration of losartan (100 mg) was estimated to be 1.2, using the maximum hepatic input total blood concentration (7.3 µmol/L). The present results show that losartan acts as a competitive inhibitor of CYP2C8-dependent drug metabolism in vitro. Subjects with a low clearance of losartan, resulting in a high average systemic blood concentration of losartan after repeated oral administration, should be carefully monitored for possible adverse reactions during co-medication with CYP2C8 substrate drugs.
CYP2C8 is a major human hepatic CYP, constituting about 7% of total microsomal CYP content in the liver, in which it catalyzes the oxidative metabolism of at least 5% of drugs.1–6) Paclitaxel, a frequently used anticancer drug, is mainly metabolized to 6α-hydroxypaclitaxel by CYP2C8 and is also used as a CYP2C8 marker substrate.1,2) Plasma concentration and area under the concentration–time curve (AUC) of paclitaxel are correlated with degree of neurotoxicity.7) Therefore, it is important to investigate the interaction between paclitaxel and possible co-medication with CYP2C8 inhibitors in order to avoid adverse events.
Losartan, an angiotensin II receptor blocker, is a substrate of both CYP2C9 and CYP3A4, although CYP2C9 appears to be more important.8,9) The maximum radioactivity in the liver was about 20–40 fold higher than that in the plasma after oral administration of 14[C]-losartan in rats, indicating that losartan accumulates to the liver.10) Losartan has been reported to have inhibitory effects on CYP2C911) and to inhibit amodiaquine metabolism by CYP2C8,12) although it is not a substrate of CYP2C8. Therefore, losartan would interact with other drugs metabolized by CYP2C8, such as paclitaxel, in the human liver. To evaluate the inhibitory effects of CYP2C8 by losartan, it is important to explore the mechanisms of CYP2C8 inhibition and its inhibitory effects on other substrates of this enzyme.
The aims of the present study were to investigate the type of CYP2C8 inhibition by losartan in relation to the magnitude of the inhibition and to estimate its apparent Ki value. Degree of inhibition after oral administration of losartan was estimated using losartan concentration in clinical settings and Ki value, in accordance with the method of Ito et al.13)
Losartan potassium was purchased from Sigma-Aldrich (St. Louis, MO, U.S.A.). Paclitaxel was purchased from Wako Pure Chemical Industries, Ltd. (Osaka, Japan). Docetaxel trihydrate used as an internal standard (IS) was purchased from Toronto Research Chemicals (North York, Canada). Human lymphoblast-expressed human CYP2C8 (rCYP2C8: 92 pmol P450/mg protein), control microsomes derived from human lymphoblast cells containing vector, and reduced nicotinamide adenine dinucleotide phosphate (NADPH) regenerating system solutions A and B were purchased from BD Gentest (Woburn, MA, U.S.A.). All other chemicals and solvents were HPLC-grade or special-grade reagents.
The following stock solutions of each compound were prepared and stored at −20°C: losartan (1 mmol/L), paclitaxel (4 mmol/L) and docetaxel (0.2 mg/mL). Paclitaxel stock solution was diluted with methanol to 0.75 mmol/L and docetaxel stock solution was diluted with acetonitrile to 10 µg/mL before use.
Inhibition Study in Human Lymphoblast-Expressed Human CYP2C8Incubation was performed using the method of Crespi et al.14) with some modifications. All incubations were carried out in the linear range of paclitaxel metabolism with respect to protein concentration (0.5–1.0 mg protein/mL), incubation time (30–120 min), and substrate concentration (7.5 µmol/L: reported Km value15)), and these conditions were consistent with recommended conditions in the package insert for rCYP2C8. Losartan concentrations in incubates (100 µL) were 0, 100, 500 and 1000 µmol/L. Incubation mixtures consisting of 83 µL of 50 mmol/L phosphate buffer (pH 7.4), 10 µL of rCYP2C8 (10 mg protein/mL: 920 pmol P450/mL) or control microsomes (10 mg protein/mL), and 6 µL of NADPH regenerating system solution (final concentration: 1.3 mmol/L NADP+, 3.3 mmol/L glucose-6-phosphate, 3.3 mmol/L MgCl2, 0.4 U/mL glucose-6-phosphate dehydrogenase) was preincubated for 3 min at 37°C. The reaction was started by adding 1 µL of 0.75 mmol/L paclitaxel dissolved in methanol. Paclitaxel concentration in the incubation mixture was 7.5 µmol/L. Methanol content in the incubation mixture was less than 1% to prevent inactivation of rCYP2C8.16) After incubation for 120 min at 37°C, the reaction was stopped by adding 30 µL of ice-cold acetonitrile. Paclitaxel was stable in the mixture within the incubation experiment (data not shown). All incubations were performed in duplicate.
HPLC AssayThe instrument was a C-R4AX chromatopac data acquisition system equipped with a Shimadzu LC-6A pump, and a SCV-6B system controller (Shimadzu Corp., Kyoto, Japan) coupled with a UV-VIS detector (Shimadzu SPD-10A VP). The column was LichroCART ODS with a particle size of 5 µm (250 mm×4 mm, Merck, Darmstadt, Germany).
Residual concentrations of paclitaxel were measured using the method of Andreeva et al.17) with some modifications. Briefly, column temperature and detection wavelength were set at 35°C and 227 nm, respectively. Mobile phase consisted of water and acetonitrile (8 : 7, v/v), and flow rate was 1.2 mL/min. Internal standard solution (10 µg/mL docetaxel; 100 µL) was added to 50 µL of incubated samples, followed by centrifuged at 6490×g for 5 min and 100 µL of supernatant was evaporated to dryness. Evaporated samples were reconstituted with 100 µL of mobile phase and were then centrifuged at 6490×g for 5 min. Supernatant (25 µL) was injected into HPLC. Under these conditions, retention times of paclitaxel and docetaxel (IS) were 11.8 min and 10.1 min, respectively.
Apparent Ki Value and Type of InhibitionThe type of inhibition and apparent Ki value for losartan on paclitaxel metabolism were determined from Dixon plots18) and Cornish–Bowden plots19) using four losartan concentrations (0, 50, 100 and 250 µmol/L). Three paclitaxel concentrations were examined at Km=7.5 µmol/L, Km/2=3.75 µmol/L and 2Km=15 µmol/L.15) All measurements were performed in duplicate.
Data AnalysisAll calculations were performed using Microsoft Excel 2011 (Version 14.3.7). Products of paclitaxel metabolite were calculated using Eq. 1.
 | (1) |
Percentage of paclitaxel metabolized (%) was calculated using Eq. 2.
 | (2) |
Metabolic activity of paclitaxel (v) was calculated using Eq. 3.
 | (3) |
In order to estimate the degree of inhibition after oral administration of losartan, Eqs. 4–6 were used.13)
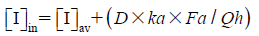 | (4) |
where [I]in is the maximum hepatic input total blood concentration, indicating the sum of the hepatic artery and portal vein concentrations during the absorption process, and [I]av is average systemic total blood concentration of losartan after repeated oral administration. D is the oral dose of losartan (100 mg), ka is the first-order absorption rate constant, Fa is the fraction of the oral dose absorbed, and Qh is the hepatic blood flow. To avoid false negative results, values of 0.1 min−1, 1.0, and 1610 mL/min were used for ka, Fa and Qh.13) [I]av was obtained by Eq. 5.
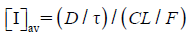 | (5) |
where τ is dose interval, CL is total clearance, and F is bioavailability. Values of 558.7 mL/min and 36.5% obtained from six healthy male subjects were used for CL and F, respectively.20) Consequently, [I]av in plasma was calculated to be 0.098 µmol/L, and blood concentration was estimated to be 0.052 µmol/L after multiplying by 0.53, which was the reported value of blood to plasma concentration ratio.20)
The degree of inhibition (R) after oral administration of losartan was calculated by Eq. 6, based on the assumption that substrate concentration was much lower than its Km value.
 | (6) |
The R statistical package (ver. 2.8.0) was used for statistical analysis.21) Williams’ multiple comparison was employed to compare the inhibitory effects in each losartan concentration against 0 µmol/L losartan.22) The p values of less than 0.05 were considered to indicate statistical significance.
Losartan inhibited paclitaxel metabolism in rCYP2C8 at all concentrations studied (p<0.05; Fig. 1). Percentage of paclitaxel metabolized with 100 µmol/L of losartan was calculated to be 42.5% of that without losartan.
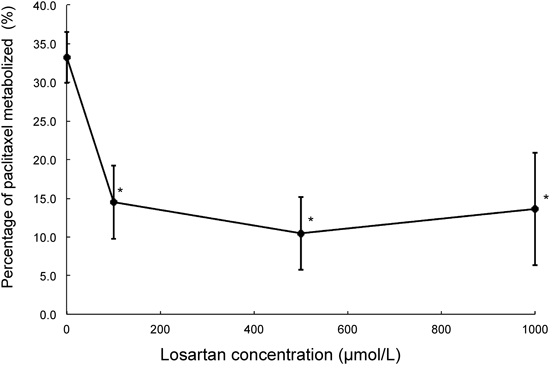
Data are expressed as means±S.D. (n=3). * p<0.05 vs. 0 µmol/L by Williams’ multiple comparison. Vertical axis shows percentage of paclitaxel metabolized by rCYP2C8, and horizontal axis shows losartan concentration.
The Dixon and the Cornish–Bowden plots of paclitaxel metabolism at different concentrations of losartan are shown in Figs. 2 and 3, respectively. Apparent Ki was estimated to be 40.7 µmol/L from the Dixon plot (Fig. 2). It was concluded from these plots that the type of CYP2C8 inhibition by losartan was competitive (Figs. 2, 3). Losartan at 50 µmol/L inhibited paclitaxel metabolism by about 60% (paclitaxel concentration: 7.5 µmol/L, Fig. 4). The R value was calculated to be 1.2, using values of 40.7 µmol/L and 7.3 µmol/L for Ki and [I]in, respectively.
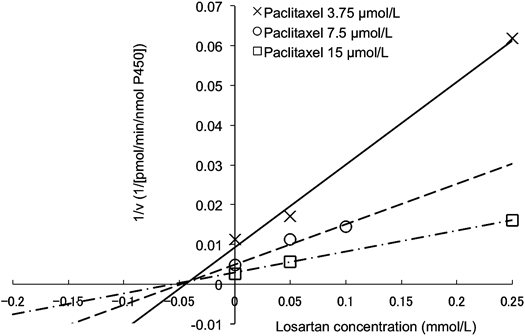
v: Metabolic activity of paclitaxel, calculated using Eq. 3 (see Materials and Methods). Solid line, dashed line, and dash–dot line show data at paclitaxel concentrations of 3.75, 7.5 and 15 µmol/L, respectively (n=1–2).
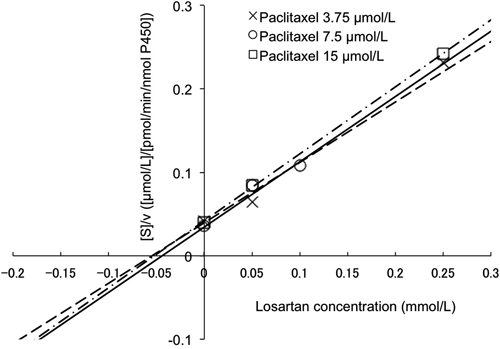
v: Metabolic activities of paclitaxel, calculated using Eq. 3 (see Materials and Methods). [S]: Paclitaxel concentrations in incubated samples. Solid line, dashed line and dash–dot line show data for paclitaxel concentrations of 3.75, 7.5 and 15 µmol/L, respectively (n=1–2).
Losartan inhibited CYP2C8-dependent paclitaxel metabolism, and this is consistent with previous reports on the inhibitory effects of losartan on amodiaquine metabolism via CYP2C8.12) Thus, losartan could interact with other substrates of CYP2C8. In the present study, 50 µmol/L losartan inhibited paclitaxel metabolism by approximately 60% (Fig. 4). Losartan would significantly inhibit paclitaxel metabolism, even at concentrations below 50 µmol/L. In this study, we used the substrate depletion assay for CYP2C8 activity using rCYP2C8. The results include the depletion of substrate by non-enzymatic degradation and adhesion to microsomal protein of substrate, despite these effects being eliminated by the use of control microsomes in parallel with test samples (Eq. 1).

Data were obtained from Dixon and Cornish–Bowden plots (Figs. 2, 3) at 7.5 µmol/L paclitaxel (n=2). Data are expressed as means±S.D.
Dixon and Cornish–Bowden plots18,19) were employed to classify the inhibition processes. The intersection point was observed above the x-axis in the Dixon plot (Fig. 2), whose x-coordinate corresponds to Ki value (dissociation constant of enzyme–inhibitor complex), indicating that the type of inhibition by losartan was competitive or mixed inhibition. On the other hand, lines were parallel on the Cornish–Bowden plot, indicating that Ki′ value (dissociation constant of enzyme–substrate–inhibitor complex) was not observed. Thus, the type of inhibition by losartan was considered to be competitive.19) This finding and the fact that losartan is not metabolized by CYP2C88) suggest that the mechanism-based inhibition of CYP2C8 by losartan is unlikely to be involved.
Apparent Ki value was estimated to be 40.7 µmol/L from the Dixon plot (Fig. 2). The R value of losartan for CYP2C8 inhibition was calculated to be 1.2 using the maximum hepatic input total blood concentration, which is assumed the highest concentration taken into the human liver, in order to avoid false negative results.13) Because this assumption was based on the physiological model, analyses were performed using the blood concentration of losartan, which is calculated by multiplying the blood–plasma concentration ratio by the plasma concentration of losartan. Some reports have used unbound inhibitor concentration for predicting increased AUC ratio in the presence of inhibitor, and it is assumed that inhibitor is transported into the liver by passive diffusion.13,23) However, losartan accumulated at high levels in the liver, at concentrations 20–40 fold higher than in plasma.10) In addition, telmisartan and valsartan, which have similar chemical structures as losartan, have been reported to be substrates of organic anion transporters that transport drugs into the liver.24,25) These findings suggest that losartan is a substrate for uptake transporters into the liver. Therefore, the total inhibitor concentration of losartan was employed for a series of analyses in the present study. CYP2C9, a key enzyme in losartan metabolism, has several genetic polymorphisms with reduced clearance of losartan in vitro and in vivo.9,26) In subjects with reduced losartan clearance, [I]av of losartan is considered to be higher than 0.052 µmol/L, indicating that the R value calculated by parameters derived from these populations is higher than 1.2. In relation to the parameters used in this study, none of the information on the genotype of CYP2C9 was indicated.20) Thus, the impact of CYP2C9 genotype should be investigated in order to elucidate the inhibitory effects of losartan on paclitaxel metabolism in vivo.
Our results showed that the interaction between losartan and paclitaxel in vivo should not be excluded, because the R value was higher than 1.1.27) However, precise prediction of the degree of inhibition is difficult in overall paclitaxel metabolism by losartan in vivo, as paclitaxel is also metabolized by CYP3A4, and its contribution to paclitaxel metabolism has large inter-individual differences.28) Further studies taking into account both the metabolic pathways of paclitaxel in human liver microsomes are required in order to assess the clinical relevance of losartan–paclitaxel interactions in vivo.
In summary, losartan has a dose-dependent inhibitory effect on CYP2C8-dependent paclitaxel metabolism. Our results demonstrate that the type of CYP2C8 inhibition by losartan is competitive. The R value for CYP2C8 inhibition after oral administration of losartan (100 mg) was estimated to be 1.2, using the maximum hepatic input total blood concentration. The present study suggests that subjects with low clearance of losartan, resulting high average systemic blood total concentration of losartan after repeated oral administration, should be carefully monitored for possible adverse reactions during co-medication with paclitaxel and/or other CYP2C8 substrate drugs.