Notes
Preparation of Antibodies and Development of an Enzyme-Linked Immunosorbent Assay for the Tyrosine Kinase Inhibitors Lapatinib and Nilotinib
2015 Volume 38 Issue 10 Pages 1652-1657
Details
2015 Volume 38 Issue 10 Pages 1652-1657
In this paper, we describe the production of the first specific antibodies against the tyrosine kinase inhibitors lapatinib and nilotinib. Anti-lapatinib antibody was obtained by immunizing rabbits with an antigen conjugated with bovine serum albumin using 3-chloro-4-((3-fluorobenzyl)oxy)aniline. Anti-nilotinib antibody was produced by immunizing mice with an antigen conjugated with bovine serum albumin using 2-(5-amino-2-methylanilino)-4-(3-pyridyl)pyrimidine. The generated antibodies were used to develop highly sensitive and specific enzyme-linked immunosorbent assays (ELISAs) for lapatinib and nilotinib in human serum. The assays were capable of detecting lapatinib and nilotinib at serum concentrations as low as 40 and 8 ng/mL, respectively. Using the two ELISAs, drugs levels were easily measured in the serum of rats after a single dose oral administration of lapatinib or nilotinib. The assays are therefore expected be valuable tools for therapeutic drug monitoring in the clinical setting and pharmacokinetic studies of lapatinib and nilotinib.
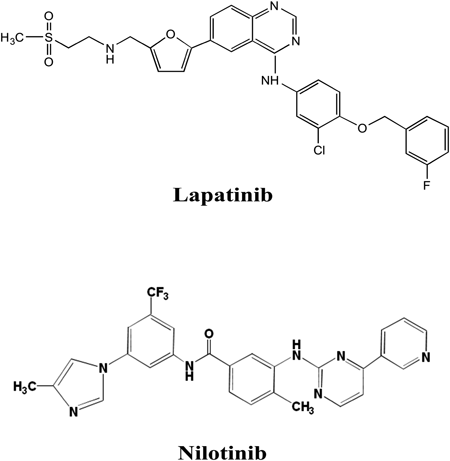
Small-molecule tyrosine kinase inhibitors (TKIs) are a class of targeted drugs for the treatment of malignant pathologies, and have widely been used in advanced cancer. Among TKIs, lapatinib is a dual inhibitor of the tyrosine kinase functionality of the epidermal growth factor receptor (EGFR or ErbB1) and HER-2/neu (erbB2) receptors of the epidermal growth factor (EGF) family,1) and has been approved for use in breast cancer treatment.2) In addition, nilotinib is a Bcr-Abl TKI that is effective in imatinib-resistant or -intolerant patients with Philadelphia-positive chronic myelogenous leukemia.3,4) Despite their proven benefit in cancer treatment, lapatinib and nilotinib exhibit inter-individual pharmacokinetic variability, and may have drug interactions and toxic effects. Therefore, to prevent treatment failure and avoid potential toxicity with TKIs, therapeutic drug monitoring (TDM) on an individual patient basis has been proposed as an approach to reduce drug toxicity and resistance, and to achieve a high level of adherence for a higher likelihood of treatment response. Current analytical methods for TKIs include high-performance liquid chromatography (HPLC)5) and liquid chromatography with tandem mass spectrometry (LC-MS/MS).6,7) Although these methods are highly sensitive and specific, they require expensive equipment and complicated sample pretreatment processes, including purification, concentration, and/or derivatization steps. Thus, the development of a simple quantification method for TKIs is needed to facilitate TDM in the clinical setting. An attractive approach is the use of enzyme-linked immunosorbent assay (ELISA), which is sensitive, specific, and has the added benefits of simplicity, low cost, and capacity for high throughput. Recently, we developed ELISAs for several TKIs and demonstrated that the assays were sensitive, specific and useful for TDM and pharmacokinetic studies of the target drugs.8,9)
This study describes a methodology for the production of antibodies against lapatinib and nilotinib. In addition, a method for the labeling of the two drugs with horseradish peroxidase (HRP) as a tracer, characterization of the specificity of the antibodies, and the measurement of lapatinib and nilotinib by ELISA is presented.
Lapatinib and nilotinib were obtained from AdooQ BioScience LLC (CA, U.S.A.). 3-Chloro-4-{(3-fluorobenzyl)oxy}aniline (CFBA) was purchased from Ark Pharm, Inc. (Libertyville, U.S.A.). 2-(5-Amino-2-methylanilino)-4-(3-pyridyl)pyrimidine (AMPP) was obtained from Tokyo Chemical Industry Co., Ltd. (Tokyo, Japan). HRP and 3,3′,5,5′-tetramethylbenzidine (TMB) were purchased from Boehringer Ingelheim Pharma GmbH (Ingelheim, Germany). All other reagents and solvents were of the highest grade commercially available.
Preparation of Immunogen for LapatinibLapatinib immunogen was prepared using a partial structure of lapatinib (CFBA) as shown in Figs. 1 and 2. CFBA (10 mg, 40 µmol) in 100 µL dimethylformamide (DMF) was acidified by the addition of 100 µL of 1 M acetic acid and was then diazotized with sodium nitrite (2.8 mg, 40.5 µmol) in 50 µL distilled water at 0°C for 10 min. The resulting solution was then mixed with bovine serum albumin (BSA) (20 mg) in 4 mL of 0.1 M borate buffer (pH 9.0), followed by a 1-h incubation at room temperature. The reaction mixture was dialyzed successively for 48 h against 50 and 1 mM phosphate buffer (pH 7.0) and H2O. The purified conjugate was lyophilized and used as an immunogen for lapatinib.
Preparation of Lapatinib AntibodyAn aliquot of a solution containing 1 mg CFBA–BSA conjugate (1.0 mL) was emulsified with an equal volume of Freund’s complete adjuvant. Two female white rabbits (Kyudo Exp. Animals, Kumamoto, Japan) were administered multiple subcutaneous injections of the solution at bilateral sites along the back. Booster injections were administered three times at biweekly intervals, using half the dose of the first immunization. The rabbits were bled from an ear vein 10 weeks after the initial immunization. The collected sera (5 mL) were separated by centrifugation at 1048×g at 4°C for 10 min and then heated at 55°C for 30 min. Fractions of immunoglobulin G (IgG) were purified using a HiTrap Protein G column (GE Healthcare, Stockholm, Sweden), with 20 mM sodium phosphate (pH 7.0) as binding buffer and 0.1 M glycine HCl (pH 2.7) as elution buffer, following the manufacturer’s protocol. The fraction that passed through the column was lyophilized and used as anti-lapatinib antibody for ELISA. The experimental protocol was approved by the Ethies Review Committee for Animal Experimentation of Sojo University.
Preparation of CFBA–HRP ConjugateEnzyme labeling of lapatinib with HRP similarly performed using CFBA (Fig. 2). A solution of CFBA (10 mg, 40 µmol) and succinic anhydride (4.0 mg, 40 µmol) in pyridine (0.5 mL) was stirred overnight at 60°C. After the addition of 1 mL water, the resulting mixture was extracted with ethylacetate and then dried over anhydrous sodium sulfate. After removing the solvent by evaporation, the obtained residue, carboxylic-modified CFBA, was dissolved in 95% dioxane (1.0 mL) and was then added to 1-ethyl-3,3-dimethylaminopropyl carbodiimide hydrochloride (EDPC) (15.2 mg, 80 µmol) and N-hydroxysuccinimide (9.2 mg, 80 µmol). The resulting solution was allowed to stand at room temperature for 2 h. A 30-µL aliquot of this reaction mixture containing succinimidyl CFBA was added directly to HRP (0.5 mg, 12.5 nmol) in 0.5 mL of 0.1 M phosphate buffer (pH 7.0), followed by a 1-h incubation at room temperature. The mixture was chromatographed on a column of Sephadex G-75 (2.0×30 cm) using 20 mM phosphate buffer (pH 7.0) containing 0.15 M NaCl and 0.1% BSA (buffer A) to remove any remaining small molecules. Three-milliliter fractions were collected and fractions 12 to 14, corresponding to the main peaks showing enzyme activity, were combined and used as a label for ELISA.
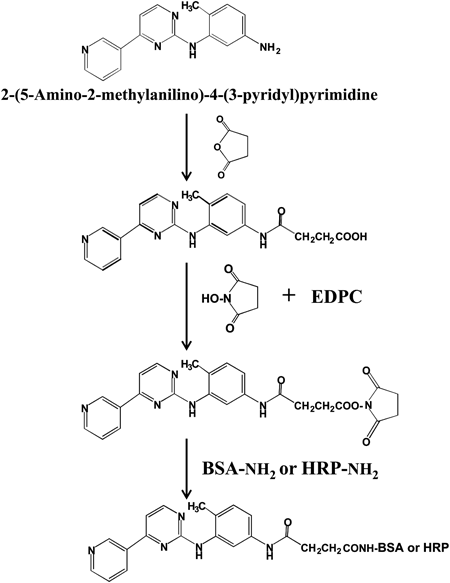
Preparation of Immunogen for NilotinibNilotinib immunogen was prepared using a partial structure of nilotinib (AMPP) as shown in Figs. 1 and 3. A solution of AMPP (10 mg, 36 µmol) and succinic anhydride (3.6 mg, 36 µmol) in pyridine (0.5 mL) was stirred overnight at 60°C. After removing pyridine by passing nitrogen through the reaction mixture, the obtained residue, carboxylic-modified AMPP, was dissolved in DMF (100 µL) and 95% dioxane (2.0 mL) and then added to EDPC (13.8 mg, 72 µmol) and N-hydroxysuccinimide (8.3 mg, 72 µmol). The resulting solution was allowed to stand at room temperature for 2 h. The reaction mixture, containing succinimidyl AMPP, was then mixed with BSA (20 mg) in 1 mL of 0.1 M phosphate buffer (pH 7.0) and further incubated at room temperature for 2 h. The reaction mixture was dialyzed successively for 48 h against 50 and 1 mM phosphate buffer (pH 7.0) and H2O. The purified conjugate was lyophilized and used as an immunogen for nilotinib.
Preparation of Nilotinib AntibodySixty-five 5-week-old female BALB/c mice (Kyudo Exp. Animals, Kumamoto, Japan) were injected intraperitoneally with 0.1 mg AMPP–BSA conjugate emulsified in complete Freund’s adjuvant. The mice were given 3 injections of 0.05 mg conjugate alone at 2-week intervals. Seven days after the final injection, the mice were euthanized and sera were collected, separated by centrifugation, heated at 55°C for 30 min, and then stored at −30°C. The obtained anti-nilotinib serum was directly used as anti-nilotinib antibody for ELISA. The experimental protocol was approved by the Ethies Review Committee for Animal Experimentation of Sojo University.
Preparation of AMPP–HRP ConjugateAMPP was labeled with HRP using a similar method as used for the preparation of nilotinib immunogen (Fig. 3). Briefly, a solution of AMPP (5 mg, 18 µmol) and succinic anhydride (1.8 mg, 18 µmol) in pyridine (0.5 mL) was stirred overnight at 60°C. After removing pyridine by passing nitrogen through the reaction mixture, the obtained residue, carboxylic-modified AMPP, was dissolved in DMF (100 µL) and 95% dioxane (1.0 mL). After the addition of 1 mL water, the resulting mixture was extracted with ethylacetate and then added to EDPC (8.0 mg, 36 µmol) and N-hydroxysuccinimide (4.15 mg, 36 µmol). The resulting solution was allowed to stand at room temperature for 2 h, and a 140-µL aliquot of the reaction mixture, containing succinimidyl AMPP, was then added directly to HRP (0.5 mg, 12.5 nmol) in 1.0 mL of 0.1 M phosphate buffer (pH 7.0), followed by a further 1-h incubation at room temperature. The mixture was chromatographed on a column of Sephadex G-75 (2.0×30 cm) using buffer A to remove any remaining small molecules. Three-milliliter fractions were collected and fractions 12 to 14, corresponding to the main peaks showing enzyme activity, were combined and used as a label for ELISA.
ELISA for Lapatinib and NilotinibELISA is based on the principle of competition between enzyme-labeled and unlabeled drugs for an immobilized antibody, followed by measurement of the marker enzyme activity of the immunocomplex bound to the solid phase. To develop an ELISA for the measurement of lapatinib and nilotinib levels in serum, the wells of microtiter plates (Nunc F Immunoplates I; Nunc, Reskilde, Denmark) were coated by loading 100 µL of anti-lapatinib antibody (2 µg/mL) or anti-nilotinib serum diluted 1 : 5000 in 10 mM Tris–HCl buffer (pH 8.5) containing 10 mM NaCl and 10 mM NaN3. After a 1-h incubation at 37°C, the wells were washed twice with buffer A, and were then incubated with 100 µL of 10 mM Tris–HCl buffer (pH 8.5) containing 10 mM NaCl, 10 mM NaN3, and 1% skim milk for 30 min at 37°C to prevent nonspecific adsorption. The anti-TKI antibody-coated wells were then filled with 25 µL of human serum samples or buffer A as a control, followed immediately by the addition of 75 µL of the pooled TKI–HRP conjugate (diluted 1 : 100 and 1 : 200 in buffer A for lapatinib and nilotinib, respectively). The plates were incubated for 3 h at 37°C and the wells were then washed thoroughly with buffer A.
The activity of the enzyme conjugate bound to each well was measured by the addition of 100 µL of 0.42 mM TMB in 0.05 M acetate–citric acid buffer (pH 5.5) containing 3% dimethyl sulfoxide and 0.01% hydrogen peroxide to each well, followed by incubation of the plates at 37°C for a suitable period for approximately 10–30 min. The enzyme reaction was stopped by the addition of 100 µL of 2.0 M H2SO4 to each well, and color intensity was then measured spectrophotometrically at 450 nm using an ELISA analyzer (ImmunoMini NJ-2300, Nalge Nunc Int. Co., Ltd., Tokyo, Japan). Concentrations were calculated from standard curves using semi-logarithmic graph paper.
Pharmacokinetic EvaluationTwo male Wistar rats (Kyudo Exp. Animals, Kumamoto, Japan) with a weight range of 180–200 g were used in this study. Oral formulations were prepared in suspension form by triturating an accurately weighed amount of powdered compound in methyl cellulose (0.5%, w/v water) in a gravimetric dilution pattern. Oral doses of 10 mg/kg (lapatinib) and 20 mg/kg (nilotinib) were administered using a gavage needle at 5 mL/kg to rats after an overnight fast (12 h). Blood samples were collected from cervical vein at 0.5, 1, 2, 3, 4, 6, 8, 12 and 24 h after post-administration, and the serum was stored at −30°C until assayed for TKIs concentrations. The serum samples were diluted 1 to 100 fold with buffer A to obtain TKIs concentrations appropriate for measurement by the two ELISAs. The experimental protocol was approved by the Ethics Review Committee for Animal Experimentation of Sojo University.
The area under the plasma concentration–time curve (AUC) of nilotinib and lapatinib is increased after the administration with meal.10,11) Similar AUC reductions are observed in lapatinib and nilotinib when co-administered with antacid drugs.12,13) Thus, the blood concentration of these TKIs is altered differently under the meal and antacid drugs administration. Recently, Giles et al. reported a correlation between hyperbilirubinemia and nilotinib trough concentrations in patients treated with nilotinib.14) Therefore, evaluation of blood these TKIs levels in patients will become a useful tool for achieving the optimum therapeutic level for patients who have experienced drug interactions or adverse side effects and for those who require dose adjustment.
In this study, we aimed to produce specific antibodies against two TKIs, lapatinib and nilotinib. To our knowledge, there is no report describing specific antibody production against two TKIs. Chemical structures of the two TKIs are shown in Fig. 1. One likely reason for the difficulty in generating antibodies against two TKIs is that they do not have a suitable reactive structure for preparing immunogen, such as a drug–BSA conjugate. Therefore, here, immunogens of lapatinib and nilotinib were prepared using partial structures of each TKI.
Anti-lapatinib antibody was obtained by immunizing rabbits with an antigen conjugated with BSA using CFBA, which was coupled by diazotization to tyrosine and histidine residues on the carrier protein (Fig. 2). The CFBA–BSA conjugate induced the formation of specific antibodies in both of the two immunized rabbits. We isolated anti-lapatinib IgG from the obtained antiserum, and confirmed that the antibodies effectively adsorbed to the wells of microtiter plates. However, no useful solid phase antibodies were detected in non-purified anti-lapatinib serum.
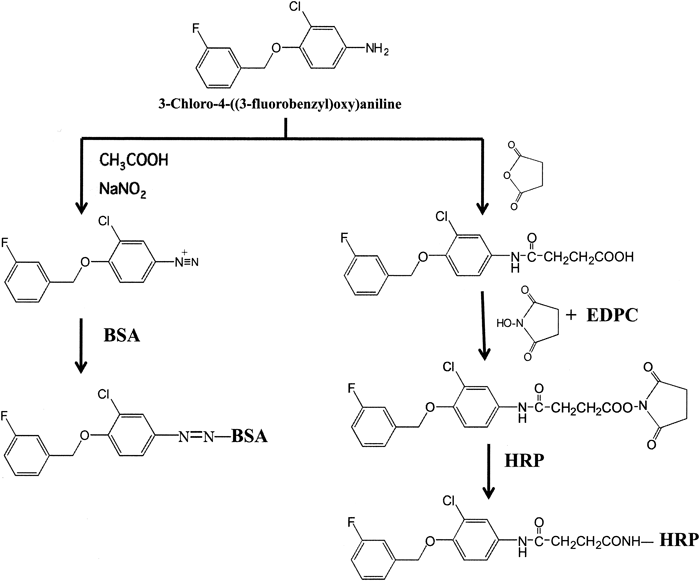
Lapatinib conjugated to HRP as a tracer was prepared using carboxylic-modified CFBA, by the hydroxysuccinimide ester method15) to eliminate the production of antibodies targeting the cross-linkage region of HRP (Fig. 2). The obtained conjugate was stable for over 6 months in elution buffer (pH 7.0) at 4°C without any loss of enzyme or immunoreactive activity.
The dose–response standard curve for lapatinib in serum is shown in Fig. 4. The curve was nearly linear on a semilogarithmic plot between 1.6 and 5000 ng/mL. For practicality, the working range of the assay was arbitrarily set between 40 and 5000 ng/mL based on the precision and accuracy of the ELISA measurements for serum levels of lapatinib (data not shown). The detection limit of lapatinib in the ELISA was 40 ng/mL (Student’s t-test, n=3, p<0.01 compared with the B0 value). As the therapeutic plasma concentration range of lapatinib is approximately 550–1710 ng/mL,16) this ELISA may be sufficiently sensitive to quantify lapatinib in TDM and pharmacokinetic studies.
The specificity of the purified anti-lapatinib antibody was evaluated based on the displacement of bound CFBA–HRP by similar compounds. Values of cross-reactivity were defined as the ratio of the test compound to CFBA for the concentration required for 50% inhibition of CFBA–HRP binding to anti-lapatinib antibody. The anti-lapatinib antibody showed 100.0% cross-reactivity with lapatinib. No detectable cross-reactivity, however, was found with m-fluorobenzyl alcohol or 4-amino-2-chlorophenol (Table 1). These findings suggest that the anti-lapatinib antibody recognize nearly the entire structure of CFBA used as a hapten, and thus are sufficiently specific to the structure of lapatinib. Lapatinib is metabolized mainly by P450 3A4 to form O- and N-dealkylated metabolites.17) The cross-reactivity of these metabolites with the anti-lapatinib antibody has not yet been confirmed. However, based on the observed specificity of the anti-lapatinib antibody, the O-dealkylated metabolite would likely show only limited cross-reactivity. In contrast, the N-dealkylated metabolite is expected to display similar cross-reactivity to that of lapatinib. However, as the maximal concentration of this metabolite detected in human plasma was relatively low,18) this ELISA may be sufficiently specific to quantify lapatinib for pharmacokinetic studies in humans.
Anti-nilotinib antibody was obtained by immunizing mice with an antigen conjugated with BSA using carboxylic-modified AMPP (Fig. 3), which was coupled to amino groups on the carrier protein by the hydroxysuccinimide ester method.15) The AMPP–BSA conjugate induced the formation of specific antibodies in each of the five immunized mice. The obtained anti-nilotinib serum, containing anti-nilotinib IgG, was directly adsorbed to the wells in microtiter plates, because useful solid-phase antibodies were present in the non-purified serum.
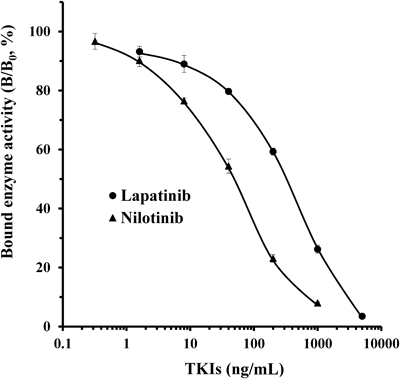
The curves show the amount (%) of bound enzyme activity for various doses of each drug (B) as a ratio of that bound by HRP-labeled drug alone (B0). ●, lapatinib; ▲, nilotinib. Each point represents the mean±S.D. of 3 replicates.
| Compounds | Cross-reactivity (%) |
|---|---|
Lapatinib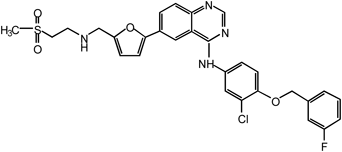 | 100 |
CFBA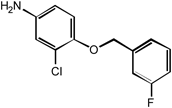 | 100 |
m-Fluorobenzyl alcohol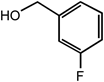 | <0.3 |
4-Amino-2-chlorophenol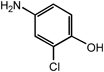 | <0.3 |
An AMPP–HRP conjugate was also prepared by the same procedure used for the nilotinib immunogen. The obtained conjugate was stable for greater than 6 months in the elution buffer (pH 7.0) at 4°C without any loss of enzyme or immunoreactive activity.
The dose–response standard curve of nilotinib in serum is shown in Fig. 4. The curve was approximately linear on a semilogarithmic plot between 0.32 and 1000 ng/mL. For practicality, the working range of the assay was arbitrarily set between 8 and 1000 ng/mL based on the detection limit of nilotinib in the ELISA (8 ng/mL; Student’s t-test, n=3, p<0.01 compared with the B0 value), and the precision and accuracy of the ELISA measurements for serum levels of nilotinib (data not shown). The therapeutic plasma concentration range of nilotinib is between 1123 and 1595 ng/mL19); therefore, this ELISA may be sufficiently sensitive to quantify nilotinib in TDM and pharmacokinetic studies.
The anti-nilotinib antibody showed 100% cross-reactivity with nilotinib. No detectable cross-reactivity, however, was found with 4-(3-pyridyl)-2-pyrimidineamine (Table 2). These findings suggest that the anti-nilotinib antibody recognizes nearly the entire structure of AMPP used as a hapten, and thus is sufficiently specific to the structure of nilotinib. In humans, nilotinib is metabolized primarily in the liver via oxidation and hydroxylation pathways mediated by CYP3A4 enzyme.20) The cross-reactivity of these metabolites with the anti-nilotinib antibody produced here has not yet been examined. However, the maximal concentration of these metabolites detected in human plasma was relatively low, and unchanged nilotinib is the main circulating component in plasma.20) Based on these finding, this ELISA may be sufficiently specific to quantify nilotinib for pharmacokinetic studies in humans.
| Compounds | Cross-reactivity (%) |
|---|---|
Nilotinib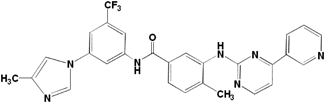 | 100 |
AMPP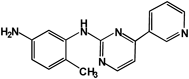 | 100 |
4-(3-Pyridyl)-2-pyrimidineamine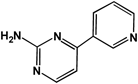 | <0.1 |
As a demonstration of the potential of the two ELISAs, preliminary pharmacokinetic studies of lapatinib and nilotinib in rats were performed (Fig. 5). Lapatinib and nilotinib were rapidly absorbed, reached peak concentrations in the serum of 650 ng/mL and 2300 ng/mL at 2 and 6 h after oral administration of lapatinib and nilotinib, respectively, and then slowly decreased. The nilotinib levels in the serum tested were almost consistent with those reported by Xia et al.21) by means of LC-MS/MS for nilotinib. Using the two ELISAs, these TKIs levels were easily measured in the serum of rats after a single dose oral administration of lapatinib or nilotinib.
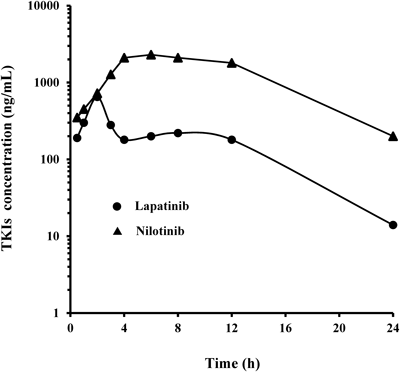
Two rats with a weight range of 180–200 g were injected with 10 mg/kg lapatinib or 20 mg/kg nilotinib. At each interval, blood samples were collected and serum levels of lapatinib and nilotinib were measured by the two ELISAs.
In conclusion, we have described the production of the first antibodies against the TKIs lapatinib and nilotinib by preparing immunogens using partial structures of each TKI. The generated antibodies against lapatinib and nilotinib were used to develop two ELISAs, which were shown to be sensitive, specific and adaptable for high-throughput analyses. The assays are therefore expected be valuable tools in studies of the TDM and pharmacokinetics of lapatinib and nilotinib.
The authors declare no conflict of interest.