Regular Articles
Orthovanadate-Induced Vasoconstriction of Rat Mesenteric Arteries Is Mediated by Rho Kinase-Dependent Inhibition of Myosin Light Chain Phosphatase
2015 Volume 38 Issue 11 Pages 1809-1816
Details
2015 Volume 38 Issue 11 Pages 1809-1816
Orthovanadate (OVA), a protein tyrosine phosphatase inhibitor, induces vasoconstriction in a Rho kinase-dependent manner. The aim of this study was to determine the mechanism underlying OVA-induced vasoconstriction of rat mesenteric arteries. OVA-induced constriction of mesenteric arterial rings treated with NG-nitro-L-arginine methyl ester (L-NAME, 0.1 mM), a nitric oxide synthase inhibitor, was significantly blocked by the Rho kinase inhibitor Y-27632 (R-(+)-trans-N-(4-pyridyl)-4-(1-aminoethyl)-cyclohexanecarboxamide, 10 µM), extracellular signal-regulated kinase 1 and 2 (Erk1/2) inhibitor FR180204 (5-(2-phenyl-pyrazolo[1,5-a]pyridin-3-yl)-1H-pyrazolo[3,4-c]pyridazin-3-ylamine, 10 µM), Erk1/2 kinase (MEK) inhibitor PD98059 (2′-amino-3′-methoxyflavone, 10 µM), epidermal growth factor receptor (EGFR) inhibitor AG1478 (4-(3-chloroanilino)-6,7-dimethoxyquinazoline, 10 µM), and Src inhibitor PP2 (4-amino-3-(4-chlorophenyl)-1-(t-butyl)-1H-pyrazolo[3,4-d]pyrimidine, 3 µM). However, the myosin light chain kinase inhibitor ML-7 (1-(5-iodonaphthalene-1-sulfonyl)-homopiperazine, 10 µM) did not affect OVA-induced constriction. Phosphorylation of myosin phosphatase target subunit 1 (MYPT1, an index of Rho kinase activity) was abrogated by inhibitors of Src, EGFR MEK, Erk1/2, and Rho kinase. OVA-stimulated Erk1/2 phosphorylation was blocked by inhibitors of EGFR, Src, MEK, and Erk1/2 but not affected by an inhibitor of Rho kinase. OVA-induced Src phosphorylation was abrogated by an Src inhibitor but not affected by inhibitors of EGFR, MEK, Erk1/2, and Rho kinase. In addition, the metalloproteinase inhibitor TAPI-0 (N-(R)-[2-(hydroxyaminocarbonyl)methyl]-4-methylpentanoyl-L-naphthylalanyl-L-alanine amide, 10 µM) and an inhibitor of heparin/epidermal growth factor binding (CRM 197, 10 µg/mL) did not affect OVA-induced contraction of rat mesenteric arterial rings. These results suggest that OVA induces vasoconstriction in rat mesenteric arteries via Src, EGFR, MEK, and Erk1/2 activation, leading to the inactivation of myosin light chain phosphatase through phosphorylation of MYPT1.
Vanadium forms numerous inorganic compounds (vanadyl sulfate, sodium metavanadate, sodium orthovanadate (OVA), vanadium pentoxide) and complexes with organic compounds.1) Because of its structural similarity to phosphate, vanadate possesses various biochemical and pharmacological properties, such as the ability to inhibit ATPases2) and protein tyrosine phosphatases (PTPases),3) epidermal growth factor (EGF)-like mitogenic activity,4) insulin-mimetic properties,5) anti-apoptotic activities,6) and antitumor or carcinogenic properties.7,8)
Vanadium compounds exert contractile effects on vascular and non-vascular smooth muscle, such as the guinea pig taenia coli, trachea, and gallbladder smooth muscle.9–11) The contractile effects of vanadates have been considered to be due to inhibition of PTPase,9,11,12) but not ATPase,13,14) because genistein, a protein tyrosine kinase inhibitor, attenuates vanadate-induced constriction. Vanadate-induced muscle constriction was regulated by activation of Rho kinase-dependent signaling in ileal longitudinal smooth muscle in guinea pigs, resulting in increased phosphorylation of the myosin light chain (MLC).15) These reports showed that vanadates inhibited the activity of protein tyrosine phosphatases, leading to Rho kinase-dependent constriction. Recently, we reported that OVA induced Rho kinase-dependent constriction and phosphorylation of the myosin phosphatase target subunit 1 (MYPT1) of myosin light chain phosphatase (MLCP). OVA-induced phosphorylation of MYPT1 was regulated by the EGF receptor (EGFR) and Src, because inhibitors of EGFR and Src abolished phosphorylation of MYPT1.16) Furthermore, metalloproteinase inhibitor TAPI-0 (N-(R)-(2-(hydroxyaminocarbonyl)methyl]-4-methylpentanoyl-L-naphthylalanyl-L-alanine amide) and an inhibitor of heparin/EGF binding abrogated OVA-induced aortic constriction and phosphorylation of EGFR and MYPT1.16) This finding indicated that OVA transactivated EGFR in vascular smooth muscle cells (VSMCs) derived from the rat thoracic aorta. These findings indicate that OVA can influence smooth muscle constriction through EGFR and Rho kinase-dependent inactivation of MLCP. Furthermore, we showed that EGF induced Ca2+ sensitization in vascular smooth muscle by Rho kinase-dependent inactivation of MLCP through the extracellular signal-regulated kinases 1 and 2 (Erk1/2) and Erk1/2 kinase (MEK) pathway.17) However, it is not clear whether OVA activates MEK and Erk1/2 signaling through the EGFR. In addition, it is unknown whether OVA induces constriction and phosphorylation of MYPT1 in mesenteric arteries in rats. Therefore, in this study, we aimed to determine whether OVA-induced constriction of rat superior mesenteric arteries is mediated by Src, EGFR, mitogen-activated protein kinase (MAPK)s, and Rho kinase.
AG1478 (4-(3-chloroanilino)-6,7-dimethoxyquinazoline), AS601245 (Z)-2-(benzo[d]thiazol-2(3H)-ylidene)-2-(2-((2-(pyridin-3-yl)ethyl)amino)pyrimidin-4-yl)acetonitrile, FR180204 (5-(2-phenyl-pyrazolo[1,5-a]pyridin-3-yl)-1H-pyrazolo[3,4-c]pyridazin-3-ylamine, L-NAME (NG-nitro-L-arginine methyl ester), ML-7 (1-(5-iodonaphthalene-1-sulfonyl)homopiperazine), PD98059 (2′-amino-3′-methoxyflavone), PP2 (4-amino-3-(4-chlorophenyl)-1-(t-butyl)-1H-pyrazolo[3,4-d]pyrimidine), SB203580 (4-(4-fluorophenyl)-2-(4-methylsulfinylphenyl)-5-(4-pyridyl)1H-imidazole), TAPI-0 (N-(R)-[2-(hydroxyaminocarbonyl)methyl]-4-methylpentanoyl-L-naphthylalanyl-L-alanine amide), and Y-27632 R-(+)-trans-N-(4-pyridyl)-4-(1-aminoethyl)-cyclohexanecarboxamide were purchased from Merck-Millipore (Tokyo, Japan). CRM 197 (an inactive diphtheria toxin) from purchased from Wako Pure Chemical Industries, Ltd. (Osaka, Japan). OVA was purchased from Nacalai Tesque (Kyoto, Japan). The inhibitor concentrations used in this study were as follows: AG1478 (10 µM), AS601245 (10 µM), CRM 197 (10 µg/mL), FR180204 (10 µM), L-NAME (0.1 mM), ML-7 (10 µM), PP2 (3 µM), PD98059 (10 µM), SB203580 (10 µM), TAPI-0 (10 µM), and Y-27632 (10 µM). The concentrations of inhibitors were selected based on our previous studies.16,17)
Organ Chamber ExperimentsAll animal experiments were carried out in accordance with the Kobe Gakuin University Experimental Animal Care Guidelines. Male Wistar rats (7–8 weeks old and 170–200 g) were anesthetized with diethyl ether. The branches of second-order superior mesenteric arteries were excised, placed in Krebs–Henseleit solution (118.4 mM NaCl, 4.7 mM KCl, 2.5 mM CaCl2, 1.2 mM KH2PO4, 1.2 mM MgSO4, 25.0 mM NaHCO3, and 11.1 mM glucose; pH 7.4), and cleaned of adherent tissue. The mesenteric arteries were cut into 2-mm rings, fixed vertically under a resting tension of 0.5 g in 5-mL organ chambers (UC-5A; Medical Kishimoto; Kyoto, Japan) filled with Krebs–Henseleit solution (37°C, pH 7.4), and aerated continuously with a gaseous mixture of 95% O2 and 5% CO2 for 60 min. Isometric tension changes were measured with a force displacement transducer (AP-5; Medical Kishimoto, Kyoto, Japan) coupled to a dual-channel chart recorder (SS-250F; SEKONIC, Tokyo, Japan). All tissue rings were exposed to 40 mM KCl for 30 min for measurement of maximal contractile force. Prior to isotonic measurements of vascular contractility, arteries were allowed to equilibrate for another 60 min. After equilibration, OVA was added to the bath solution to reach a final concentration of 0.05–5 mM. For some experiments, nitric oxide (NO) synthase (NOS) inhibitor L-NAME (0.1 mM) was added to the bath solution 30 min before the addition of OVA. The inhibitors used in the present study were dissolved in dimethyl sulfoxide and added to the organ chambers in 10-µL volumes 15 min before the addition of OVA. The contractile effect of OVA was expressed as a percentage of the maximal force evoked by 40 mM KCl.
Western BlottingThe rings of mesenteric arteries were equilibrated as described above and treated with L-NAME (0.1 mM) for 30 min, followed by treatment with OVA for 5 min. In some experiments, the inhibitors were added to the organ bath 15 min before the addition of OVA. The mesenteric arterial rings were homogenized in 50 µL lysis buffer (50 mM Tris–HCl (pH 7.4), 5 mM ethylenediaminetetraacetic acid (EDTA), 150 mM NaCl, 2 mM sodium orthovanadate, 50 mM NaF, protease inhibitor cocktail (Nacalai Tesque, Kyoto, Japan), and 0.1% nonylphenyl–polyethylene glycol). Insoluble material was removed by centrifugation at 15000×g for 10 min at 4°C. The protein concentration was determined using the BCA Protein Assay Kit from Thermo Scientific (Rockford, IL, U.S.A.). Equal amounts of protein (0.8 µg/lane) were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene fluoride (PVDF) membranes (Immobilon-P®; Millipore, Billerica, MA, U.S.A.). The blots were blocked with 5% skim milk in Tris-buffered saline (10 mM Tris in 100 µM NaCl containing 0.1% Tween 20, pH 7.5). The membranes were then incubated with primary antibodies against β-actin (1 : 1000; #4967, Cell Signaling, Danvers, MA, U.S.A.), MYPT1 (1 : 200; sc-25618, Santa Cruz Biotechnology, Santa Cruz, CA, U.S.A.), Thr-853-phosphorylated MYPT1 (1 : 200; sc-17432-R, Santa Cruz Biotechnology, Santa Cruz, CA, U.S.A.), Erk1/2 (1 : 1000; #4695-P, Cell Signaling), Thr-202/Tyr-204-phosphorylated Erk1/2 (1 : 1000; #9101-S, Cell Signaling), Src (1 : 1000; #2102-S, Cell Signaling), and Src phosphorylated at Tyr-416 (1 : 1000; #2101-S, Cell Signaling). Bound antibodies were detected with peroxidase-conjugated anti-rabbit immunoglobulin G (IgG) secondary antibodies by enhanced chemiluminescence (Chemi-Lumi One Super; Nacalai Tesque). Immunoblots were quantified using densitometry with Versa Doc 5000MP (Bio-Rad Laboratories, Hercules, CA, U.S.A.) and Quantity One software (Bio-Rad Laboratories).
Statistical AnalysisAll data are expressed as mean±standard error of the mean (S.E.M.). Statistical comparisons were performed using one-way ANOVA with pair-wise comparisons performed using the Bonferroni–Dunn method. Comparisons of concentration response curves were carried out by repeated-measures two-way ANOVA followed by the Bonferroni–Dunn method. Differences were considered significant at p<0.05.
The contractile response to cumulative doses of OVA was studied in tissue rings prepared from rat mesenteric arteries. OVA produced a weak and concentration-dependent contractile response in mesenteric arterial rings (Figs. 1A, C). The addition of NOS inhibitor L-NAME (0.1 mM) into the organ bath significantly increased the contractile response to OVA in a dose-dependent manner (Figs. 1B, C). Because OVA is a potent activator of endothelial NOS,18) it is likely that endothelium-derived NO attenuated the contractile effects of OVA on aortic smooth muscle. Because the aim of this study was to evaluate the effect of OVA on vasoconstriction of smooth muscle in mesenteric arteries, subsequent studies were carried out in L-NAME-treated mesenteric arteries.

A: left tracing shows tension changes in response to KCl (40 mM) and right tracing shows tension changes in response to the cumulative addition of OVA in the absence of L-NAME. B: representative isometric force tracings of mesenteric arterial rings in response to KCl (40 mM: left) and the cumulative addition of OVA in the presence of L-NAME (0.1 mM; added to the organ bath 30 min before OVA). C: concentration–response curves of mesenteric arterial rings in the presence (closed circle) or absence (open circle) of L-NAME (0.1 mM; added to the organ bath 30 min before OVA). The OVA concentration–response curves were constructed by measuring the force after each stepwise increase in the OVA concentration. Contractile force was expressed as a percentage of the maximal force evoked by 40 mM KCl. Data are presented as the mean±S.E.M. for mesenteric arterial rings from 4–5 animals, * p<0.05 vs. rings without L-NAME.
We studied the effects of inhibitors of Rho kinase, Src, EGFR, MEK, Erk1/2, c-Jun N terminal kinase (JNK) and p38 MAPK on OVA-induced vasoconstriction in L-NAME-treated (0.1 mM) mesenteric arterial rings (Fig. 2). The contractile effects of OVA were blocked by Rho kinase inhibitor Y-27632 (10 µM, p<0.05 vs. control, n=5, Fig. 2A), Src inhibitor PP2 (3 µM, p<0.05 vs. control, n=5, Fig. 2B), and EGFR inhibitor AG1478 (10 µM, p<0.05 vs. control, n=5, Fig. 2B). The increase in OVA-induced constriction produced by L-NAME (0.1 mM) was blocked by pretreatment with MEK inhibitor PD98059 (10 µM, p<0.05 vs. control, n=5, Fig. 2C) or Erk1/2 inhibitor FR180204 (10 µM, p<0.05 vs. control, n=5, Fig. 2C), but not by pretreatment with JNK inhibitor AS601245 (10 µM) or p38 MAPK inhibitor SB203580 (10 µM) (Fig. 2C). In contrast, myosin light chain kinase (MLCK) inhibitor ML-7 (10 µM) did not significantly affect OVA-induced constriction (Fig. 2A). Treatment with ML-7 at a concentration of 10 µM abolished phenylephrine-induced constriction in L-NAME-treated mesenteric arterial rings (data not shown).

L-NAME (0.1 mM) was added 30 min before the addition of OVA and A: ML-7 (10 µM) or Y-27632 (10 µM); B: AG1478 (10 µM) or PP2 (3 µM); C: PD98059 (10 µM), FR180204 (10 µM), SB203580 (10 µM), or AS601245 (10 µM); or D: TAPI-0 (10 µM) or CRM 197 (10 µg/mL) to the organ bath 15 min before the addition of OVA. Control rings were exposed to dimethyl sulfoxide without inhibitors. The concentration–response curves for OVA were constructed by measuring the force after each stepwise increase in the OVA concentration. Contractile force was expressed as a percentage of the maximal force evoked by 40 mM KCl. Data are expressed as the mean±S.E.M. for mesenteric arterial rings taken from 5 animals, * p<0.05 vs. control rings.
We showed that OVA-induced vasoconstriction of the thoracic aorta depends on transactivation of EGFR via shedding of pro-heparin-binding (HB)-EGF in rat thoracic aorta.16) To investigate whether OVA-induced vasoconstriction was caused by transactivation of EGFR in mesenteric arteries, we tested the effects of two inhibitors on OVA-induced vasoconstriction: TAPI-0, a metalloproteinase inhibitor (10 µM), and CRM 197, a diphtheria toxin mutant (10 µg/mL) that specifically blocks the action of pro-HB-EGF. However, neither TAPI-0 nor CRM 197 affected OVA-induced vasoconstriction in L-NAME-treated rat mesenteric arterial rings (Fig. 2D).
Effect of OVA on Phosphorylation of Src, Erk1/2, and MYPT1 in Rat Mesenteric Arterial RingsTo determine whether OVA induces activation of Src, Erk1/2, and Rho-kinase, we measured levels of Src phosphorylation at Tyr-416, Erk1/2 phosphorylation at Thr-202/Tyr-204, and MYPT1 phosphorylation at Thr-853 in rat mesenteric arterial rings by Western blotting. As shown in Fig. 3, treating mesenteric arterial rings with OVA rapidly increased the ratio of phosphorylated protein to total protein for Src, Erk1/2, and MYPT1 within 2 min of exposure, after which the increased levels of phosphorylation were maintained for 10 min. After 15 min, the levels of phosphorylated Src and Erk1/2 returned to basal levels.

L-NAME (0.1 mM) was added to the organ bath 30 min prior to the addition of OVA. The time course of changes in the phosphorylated forms of MYPT1 (Thr-853), Erk1/2 (Thr-202/Tyr-204), and Src (Tyr-416) were measured in mesenteric arterial rings after treatment with OVA (5 mM) by Western blotting. A: representative blots of phosphorylated and total MYPT1, Erk1/2, and Src. B: Bar graphs demonstrating densitometric quantification of the ratios of phosphorylated to total MYPT1, Erk1/2, and Src. Data are expressed as the mean±S.E.M. of 4 independent experiments; * p<0.05 vs. untreated rings (0 min).
To determine whether Rho-kinase is a downstream effector of Src, EGFR, MEK, Erk1/2, JNK, and/or p38 MAPK during OVA-induced vasocontraction, we measured levels of MYPT1 phosphorylation at Thr-853 as an index of Rho kinase activity19) in rat mesenteric arterial rings 5 min after treatment with OVA in the presence or absence of various protein kinase inhibitors. As shown in Fig. 4, MYPT1 phosphorylation increased by treatment with OVA (5 mM). OVA-induced phosphorylation of MYPT1 was abolished by a specific inhibitor of Rho kinase (Y-27632 (10 µM) vs. OVA, p<0.05, n=5, Fig. 4), Src (PP2 (3 µM) vs. OVA, p<0.05, n=5, Fig. 4) and EGFR (AG1478 (10 µM) vs. OVA, p<0.05, n=5, Fig. 4). Inhibitors of MEK (PD98059 (10 µM) vs. OVA, p<0.05, n=5, Fig. 4) and Erk1/2 (FR180204 (10 µM) vs. OVA, p<0.05, n=5, Fig. 4) partially and significantly inhibited OVA-induced phosphorylation of MYPT1. However, specific inhibitors of JNK (AS601245, 10 µM) and p38 MAPK (SB203580, 10 µM) did not affect OVA-induced phosphorylation of MYPT1. These results suggest that Rho kinase is a downstream target of Erk1/2, MEK, Src, and/or EGFR, which are activated by OVA in L-NAME-treated mesenteric arterial rings.
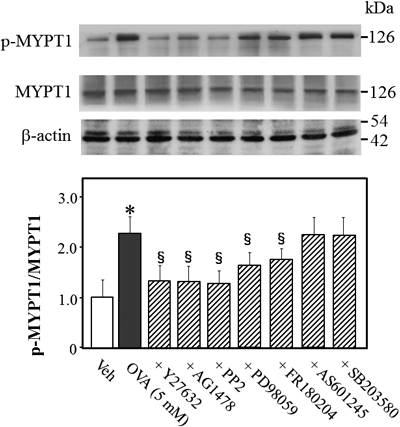
L-NAME (0.1 mM) was added to the organ bath 30 min prior to the addition of OVA. Phosphorylated (Thr-853) and total MYPT1 in mesenteric arterial rings were measured by Western blotting 5 min after treatment with OVA (5 mM). The bar graphs show the densitometric data as the ratio of phosphorylated MYPT1 to total MYPT1. The mesenteric arterial rings were exposed to Y-27632 (10 µM), AG1478 (10 µM), PP2 (3 µM), PD98059 (10 µM), FR180204 (10 µM), AS601245 (10 µM), SB203580 (10 µM), or vehicle (Veh; 10 µL dimethyl sulfoxide) 15 min prior to OVA treatment. Data are presented as the mean±S.E.M. from 4 independent experiments; * p<0.05 vs. Veh; §p<0.05 vs. OVA alone.
To determine whether MEK/Erk1/2 signaling is the downstream effector of EGFR and/or Src during OVA-induced Rho kinase activation, we measured phosphorylation of Erk1/2 at Thr-202/Tyr-204 in L-NAME-treated rat mesenteric arterial rings 5 min after treatment with OVA in the presence or absence of various inhibitors. OVA-induced phosphorylation of Erk1/2 was blocked by specific inhibitors of MEK (PD98059 (10 µM) vs. OVA, p<0.05, n=5, Fig. 5), Erk1/2 (FR180204 (10 µM) vs. OVA, p<0.05, n=5, Fig. 5), Src (PP2 (3 µM) vs. OVA, p<0.05, n=5, Fig. 5), and EGFR (AG1478 (10 µM) vs. OVA, p<0.05, n=5, Fig. 5), but not by a Rho kinase inhibitor (Fig. 5), suggesting that Erk1/2 signals upstream of Rho kinase.

L-NAME (0.1 mM) was added to the organ bath 30 min prior to the addition of OVA. Phosphorylated (Thr-202/Tyr-204) and total Erk1/2 in mesenteric arterial rings were measured by Western blotting 5 min after treatment with OVA (5 mM). The bar graphs show the densitometric data as the ratio of phosphorylated Erk1/2 to total Erk1/2. The mesenteric arterial rings were exposed to Y-27632 (10 µM), AG1478 (10 µM), PP2 (3 µM), PD98059 (10 µM), FR180204 (10 µM), or vehicle (Veh; 10 µL dimethyl sulfoxide) 15 min prior to OVA treatment. Data are presented as the mean±S.E.M. from 4 independent experiments; * p<0.05 vs. Veh; §p<0.05 vs. OVA alone.
To determine whether Src is the upstream effector of EGFR during OVA-induced Rho kinase activation, we measured levels of Src phosphorylation at Tyr-416 in L-NAME-treated rat mesenteric arterial rings 5 min after treatment with OVA in the presence or absence of various inhibitors. Src phosphorylation increased significantly after treatment with OVA. OVA-induced phosphorylation of Src was blocked by Src inhibitor (PP2 (3 µM) vs. OVA, p<0.05, n=5, Fig. 6), but not by inhibitors (10 µM) of MEK, Erk1/2, EGFR, and Rho kinase (Fig. 6), suggesting that Src signals upstream of EGFR, MEK, Erk1/2, and Rho kinase in OVA-induced contraction of mesenteric arterial rings.

L-NAME (0.1 mM) was added to the organ bath 30 min prior to the addition of OVA. Phosphorylated (Tyr-416) and total Src in mesenteric arterial rings were measured by Western blotting 5 min after treatment with OVA (5 mM). The bar graphs show the densitometric data as the ratio of phosphorylated Src to total Src. The mesenteric arterial rings were exposed to Y-27632 (10 µM), AG1478 (10 µM), PP2 (3 µM), PD98059 (10 µM), FR180204 (10 µM), or vehicle (Veh; 10 µL dimethyl sulfoxide) 15 min prior to OVA treatment. Data are presented as the mean±S.E.M. from 4 independent experiments; * p<0.05 vs. Veh; §p<0.05 vs. OVA alone.
The principal finding in the present study was that OVA-induced vasoconstriction in mesenteric arteries is mediated by Src, EGFR, MEK, Erk1/2, and Rho kinase-dependent signaling. OVA-induced phosphorylation of MYPT1 was mediated by activation of Src, EGFR, MEK, Erk1/2, and/or Rho kinase, because inhibition of these proteins abolished MYPT1 phosphorylation.
In the endothelium, tyrosine phosphatase inhibitors such as vanadates stimulate release of NO,20,21) but not of prostaglandins,21) leading to inhibition of Rho kinase signaling.22) Rho kinase inhibited phosphorylation of endothelial NOS (eNOS) at Ser1177 and decreased expression of eNOS mRNA, thereby inhibiting NO production in endothelial cells.22) These findings suggest that the Rho kinase cascade interacts with the NO signaling cascade. Therefore, to exclude the possibility that NO modified the constrictive functions of smooth muscle, we treated mesenteric arterial rings with NOS inhibitor L-NAME.
Smooth muscle constriction was regulated by the balance between the activities of MLCK and MLCP. MLCP inhibition leads to increased MLC phosphorylation and force generation without changes in cytosolic Ca2+ concentrations.23) Activation of Rho kinase leads to MYPT1 phosphorylation, resulting in suppression of MLCP activity and Ca2+-sensitization, thereby enhancing constriction without changing cytosolic Ca2+ concentrations or MLCK activity.23) Several studies show that vanadium compounds stimulate smooth muscle constriction, which is mediated by activation of Rho kinase in several tissue types.9–14,24–26) Mori and Tsushima showed that Rho kinase played a role in OVA-induced smooth muscle constriction using guinea pig ileal longitudinal smooth muscle; Rho kinase inhibitor Y-27632 blocked OVA-induced constriction and MLC phosphorylation.15) Our study in rat thoracic aorta tissue also showed that OVA-induced constriction was abolished by a Rho kinase inhibitor.16) However, it is unknown whether OVA induces constriction in mesenteric arteries via Rho kinase signaling. Here, we identified a primary role for Rho kinase in OVA-induced constriction of mesenteric arteries, because Rho kinase inhibitor Y-27632 abolished OVA-induced vasoconstriction and MYPT1 phosphorylation, whereas MLCK inhibitor ML-7 did not affect OVA-induced vasoconstriction. These studies showed that the contractile effects of OVA on smooth muscle were not accompanied by an increase in cytosolic Ca2+, but instead occurred because of increased Ca2+ sensitivity of the contractile apparatus.26–28)
Src is a nonreceptor type tyrosine kinase that activates EGFR either directly through phosphorylation at Tyr-845 in the cytoplasm29,30) or indirectly via the metalloproteinase-catalyzed formation of HB-EGF from pro-HB-EGF.31) In a previous study using rat thoracic arteries, we showed that OVA-induced vasoconstriction depended on transactivation of EGFR via shedding of pro-HB-EGF.15) OVA-induced vasoconstriction of mesenteric arterial rings was abolished by inhibitors of EGFR and Src, suggesting that Src and EGFR are involved in OVA-induced vasoconstriction. However, TAPI-0, a metalloproteinase inhibitor, and CRM 197, a diphtheria toxin mutant that specifically blocks the action of pro-HB-EGF, did not block OVA-induced vasoconstriction of mesenteric arterial rings. Therefore, it appears that OVA-induced vasoconstriction is not mediated by indirect transactivation of EGFR in rat mesenteric arteries. However, we showed in a previous study that OVA-induced vasoconstriction was dependent on EGFR transactivation in rat thoracic arteries.17) The reasons for the differences in OVA-induced constriction of mesenteric arteries and the thoracic aorta are unclear, and further studies are needed to resolve the mechanisms underlying differences in EGFR activation by OVA in different tissue types.
In mesenteric arterial rings, OVA-induced vasoconstriction and phosphorylation of MYPT1 were partially and significantly inhibited by treatment with inhibitors of Erk1/2 and MEK, indicating that OVA-induced vasoconstriction and phosphorylation of MYPT1 were regulated in part through MAPKs such as MEK and Erk1/2. MAPKs are activated by a variety of stimuli, including osmotic shock, cytokines, growth factors, and drugs. The 3 major MAPKs in mammals, Erk1/2, JNK, and p38 MAPK, are regulated by distinct signaling pathways. EGFR activation leads to activation of MAPK signaling by Erk1/2,32) JNK,33) and p38 MAPK.34) Platelet-derived growth factor-induced chemotaxis and proliferation of VSMCs were regulated by Erk1/2, because Erk1/2 inhibitor PD98059 abolished platelet-derived growth factor-induced chemotaxis and proliferation of VSMCs.35) Recently, we reported that EGF increased Ca2+ sensitization in vascular smooth muscle and stimulated phosphorylation of Erk1/2 and MYPT1.17) Chen et al. reported that a nicotinamide adenine dinucleotide phosphate (NADPH) oxidase inhibitor inhibited stretch-induced phosphorylation of Erk1/2, JNK, and p38 MAPK. Moreover, inhibitors of p38 MAPK and NADPH oxidase blocked stretch-induced vascular cell alignment.36) We also reported that p38 MAPK was involved in hyperosmotic vasoconstriction via stimulation of MLC phosphorylation and cytoskeleton reorganization through pathways independent of MLCK activation and/or Rho kinase-mediated mechanisms.37) Rho kinase activation by protein kinase C-delta and proline-rich tyrosine kinase 2 mediates angiotensin II-induced VSMC migration via JNK activation.38) Taken together, studies show that MAPK plays an important role in regulation of vascular smooth muscle function. In mesenteric arteries, we showed that inhibition of tyrosine phosphatase induced constriction via activation of MAPKs, especially MEK and Erk1/2. Because OVA-induced constriction and MLCP inactivation were partially inhibited by treatment with inhibitors of MEK and Erk1/2, OVA-induced constriction was mainly regulated by MEK and Erk1/2 signaling.
The present study demonstrated for the first time that downstream signaling by Src-activated EGFR is linked to Rho kinase-mediated MLCP inactivation, thereby regulating smooth muscle contractility in rat mesenteric arterial rings. Figure 7 illustrates the key steps in a proposed model for the mechanism through which PTPase inhibition by OVA leads to increased contractility of rat mesenteric arteries. Inhibition of unidentified PTPase(s) by vanadate activates Src, which in turn induces EGFR activation. Signaling downstream of activated EGFR stimulates Rho kinase activity, which activates MEK and Erk1/2, leading to inactivation of MLCP via MYPT1 phosphorylation and increasing mesenteric arterial contractility. OVA-induced vasoconstriction is dependent on direct transactivation of EGFR, but not dependent on indirect transactivation of EGFR, via shedding of pro-HB-EGF in rat mesenteric arterial rings. However, the reason for the difference in OVA-induced constriction between the thoracic aorta and mesenteric arteries is unclear. In addition, we could not identify the target of OVA in Src-dependent constriction. Further studies are required to comprehensively evaluate the regulation of protein tyrosine phosphatases in order to understand the mechanisms underlying vasoconstriction.

Inhibition of protein tyrosine phosphatase(s) by vanadate induces activation of Src (Tyr-416 phosphorylation), which transactivates epidermal growth factor receptor (EGFR) directly via Src-catalyzed phosphorylation of EGFR at Tyr-845. EGFR activation induces downstream signal transduction via the MEK/Erk1/2 pathway, resulting in inactivation of myosin light chain phosphatase (MLCP) via phosphorylation of myosin phosphatase target subunit 1 (MYPT1) at Thr853, which results in enhanced MLC phosphorylation via the Ca2+-dependent activation of myosin light chain kinase (MLCK).
This study was partially supported by a Grant-in-Aid for Scientific Research (C) (No. 15K07984) from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
The authors declare no conflict of interest.