Abstract
Fibroblast growth factors (Fgfs) are polypeptide growth factors with diverse biological activities. While several studies have revealed that Fgf23 plays important roles in the regulation of phosphate and vitamin D metabolism, the additional physiological roles of Fgf23 remain unclear. Although it is believed that osteoblasts/osteocytes are the main sources of Fgf23, we previously found that Fgf23 mRNA is also expressed in the mouse thymus, suggesting that it might be involved in the immune system. In this study we examined the potential roles of Fgf23 in immunological responses. Mouse serum Fgf23 levels were significantly increased following inoculation with Escherichia coli or Staphylococcus aureus or intraperitoneal injection of lipopolysaccharide. We also identified activated dendritic cells and macrophages that potentially contributed to increased serum Fgf23 levels. Nuclear factor-kappa B (NF-κB) signaling was essential for the induction of Fgf23 expression in dendritic cells in response to immunological stimuli. Moreover, we examined the effects of recombinant Fgf23 protein on immune cells in vitro. Fgfr1c, a potential receptor for Fgf23, was abundantly expressed in macrophages, suggesting that Fgf23 might be involved in signal transduction in these cells. Our data suggest that Fgf23 potentially increases the number in macrophages and induces expression of tumor necrosis factor-α (TNF-α), a proinflammatory cytokine. Collectively, these data suggest that Fgf23 might be intimately involved in inflammatory processes.
The fibroblast growth factors (Fgfs) family consists of 22 polypeptide growth factors that are widely expressed in developing and adult tissues and regulate diverse biological processes, including angiogenesis, mitogenesis, cellular differentiation, and tissue repair.1,2) Fgfs can be classified into three groups, canonical, intracellular, and hormone-like Fgfs. The canonical Fgfs bind to and activate Fgf receptors (Fgfrs) on the cell surface, resulting in the activation of several cytoplasmic signal transduction pathways. These canonical Fgfs function in a paracrine manner. The intracellular Fgfs act as intracellular signaling molecules in an Fgfr-independent manner. Hormone-like Fgfs, including Fgf15/19, Fgf21, and Fgf23, depends on Fgfrs to elicit biological responses, although they potentially function in an endocrine manner.3) One of these hormone-like Fgfs, Fgf23, was originally identified in mice and humans by a DNA database search and was a well-known physiological regulator of phosphate and vitamin D metabolisms.3–6) Fgf23 is produced by osteoblasts/osteocytes in response to 1,25-dihydroxyvitamine D, the biologically active form of vitamin D.3,5,6) Secreted Fgf23 increases urinary phosphate excretion by reducing luminal expression of sodium-phosphate co-transporters in the proximal tubule. Secreted Fgf23 also attenuates systemic levels of 1,25-dihydroxyvitamine D, which stimulates phosphate and calcium absorption from the intestinal tract, by reducing expression of 1-α hydroxylase responsible for the synthesis of the 1,25-dihydroxyvitamine D and inducing expression of 24-hydroxylase which inactivates the 1,25-dihydroxyvitamine D in kidney. Dysregulated Fgf23 signaling causes numerous human diseases, including autosomal dominant hypophosphatemic rickets (ADHR) and human tumor-induced osteomalacia (TIO).3) Thus, the current understanding is that Fgf23 is an essential regulator of phosphate and vitamin D metabolisms in mice and human. However, additional physiological roles of Fgf23 almost remain unclear.
Previously we showed by Northern blot analysis that Fgf23 is expressed in thymus of adult mice.4) In addition, Katoh and Katoh reported in silico expression analyses on Fgf23 orthologs revealing that mouse Fgf23 is expressed in dendritic cells (DCs) and activated spleen.7) These findings suggested that Fgf23 might serve potential roles in systemic immune responses. Therefore, in this report, we examined the expression and the effects of Fgf23 in mouse immune tissues and cells. We found that Fgf23 expression was induced in activated DCs and macrophages in response to immunological stimuli via nuclear factor-kappa B (NF-κB) signaling. Recombinant Fgf23 induced tumor necrosis factor-α (TNF-α) expression and increased the number of cultured macrophages. These data suggest that Fgf23 may be involved in the inflammatory process, in addition to the systemic mineral metabolisms.
MATERIALS AND METHODS
MiceFemale BALB/c mice (H-2d) were purchased from CLEA Japan Inc., and 5–8-week-old mice were used in the study. To study Fgf23 function in immunological responses, these mice were inoculated intraperitoneally with 1×108 colony forming units (CFU) of live Escherichia coli (E. coli) ATC C 11775 (bioMérieux) or 5×108 CFU or Staphylococcus aureus (S. aureus) ATC C 6538 (bioMérieux) intraperitoneally. Alternatively, mice were injected intraperitoneally with 50 µg/kg of O111 : B4 lipopolysaccharides (LPS) (Sigma). Animal care and processing were performed in accordance with the guidelines for proper conduct of animal experiments by the Science Council of Japan and were approved by the animal care committee at our institution.
Measurement of Fgf23 and TNF-α LevelsThe levels of Fgf23 protein were measured using a FGF-23 ELISA Kit according to the manufacturer’s protocols (Kinos Inc.). Although intact Fgf23 protein could be detected by this ELISA kit, the cleaved form of Fgf23 could not. TNF-α secretion into culture medium was determined using the Murine TNF-α standard enzyme-linked immunosorbent assay (ELISA) kit according to the manufacturer’s protocols (Peprotech).
Reverse Transcriptase-Quantitative Polymerase Chain Reaction (RT-qPCR) AnalysisRT-qPCR was used to examine mRNA expression levels of Fgf23 and various other cytokines. Total RNA was isolated from cultured cells or tissues using the RNeasy kit (Qiagen), and first-strand cDNA synthesis was performed using the ReverTra Ace qPCR RT kit (Toyobo). RT-qPCR was performed using Thunderbird SYBR qPCR mix (Toyobo) and primers for Fgf23, Fgf receptor 1c (Fgfr1c), interleukin-6 (IL-6), interferon-γ (IFN-γ), TNF-α, and 18S. 18S ribosomal RNA (rRNA) levels were used as an internal control. The sequences of the primers used are as the following: 18S; forward primer 5′-CGA ACG TCT GCC CTA TCA ACT T-3′ and reverse primer 5′-CCG GAA TCG AAC CCT GAT T-3′, FGF23; forward primer 5′-TGG GCA CTG CTA GAG CCT AT-3′ and reverse primer 5′-TGG CTC CTG TTA TCA CCA CA-3′, Fgfr1c; forward primer 5′-CTT GAC GTC GTG GAA CGA TCT-3′ and reverse primer 5′-TTC CAG AAC GGT CAA CCA TGC AGA-3′, IL-6; forward primer 5′-TGA TGC ACT TGC AGA AAA CA-3′ and reverse primer 5′-ACC AGA GGA AAT TTT CAA TAG GC-3′, IL-1β,; forward primer 5′-TGT GAA ATG CCA CCT TTT GA-3′ and reverse primer 5′-GGT CAA AGG TTT GGA AGC AG-3′, TNF-α; forward primer 5′-CCA CCA CGC TCT TCT GTC TAC-3′ and reverse primer 5′-AGG GTC TGG GCC ATA GAA CT-3′.
Separation of Spleen LymphocytesSpleens were removed from female BALB/c mice, pressed through a cell strainer (100 µm), and collected into a 50 mL tube in phosphate buffered saline (PBS). Subsequently, Erythrocytes were lysed in 0.38% NH4Cl–Tris buffer. Cells were washed again, resuspended in PBS and cell counts were determined. First, B cells were isolated from 108 splenocytes by a positive selection with CD19 Microbeads, MACS columns, and MACS separators according to the manufacturer’s protocols (Miltenyi Biotech). Subsequently, CD8+ splenocytes were isolated with CD8a (Ly-2) Microbeads (Miltenyi Biotech) from the residual cells depleted of B cells. Finally, CD4+ T cells were isolated from the residual cells depleted of B cells and CD8+ cells, using the CD4+ T Cell Isolation Kit 2 (Miltenyi Biotech). Over 80% of the cells expressing B220, CD8a, and CD4 were used as B cells, CD8+ cells, and CD4+ T cells, respectively. These cells were seeded at a density of 1×106 cells/mL and cultured at 37°C in RPMI 1640 medium (Gibco) containing 0.03 mg/mL L-glutamine, 100 unit/mL penicillin, and 100 µg/mL streptomycin with 10% heat-inactivated fetal bovine serum (FBS). Cells were stimulated for 3 h by culturing in RPMI 1640 supplemented with 25 ng/mL phorbol myristate acetate (PMA) (Sigma-Aldrich) and 1 µg/mL ionomycin (Enzo).
Isolation of Resident MacrophagesMurine resident peritoneal macrophages were isolated by peritoneal lavage using precooled PBS as described previously.8) Lavage fluids containing macrophages were pooled and centrifuged at 100×g for 5 min. Cells were seeded at a density of 1×106 cells/mL. After washing with RPMI 1640 medium (Gibco), adherent cells were enriched for macrophages by adherence to plastic for 2 h and cultured in RPMI 1640 medium (Gibco) with 10% heat-inactivated FBS at 37°C. To study Fgf23 induction in macrophages, cells were cultured for 3 h in RPMI 1640 medium containing 100 ng/mL LPS. Otherwise, macrophages in RPMI-1640 medium containing 2% dimethyl sulfoxide (DMSO) or 20 µg/mL furin inhibitor I (Calbiochem), were stimulated by 100 ng/mL LPS for 24 h. Thereafter, the culture supernatant was obtained to measure the Fgf23 amount by ELISA. To examine the effects of Fgf23 protein on macrophages, macrophages were plated at a density of 1×106 cells/mL. After washing with RPMI 1640 medium, adherent cells were enriched by plastic adherence for 2 h in RPMI 1640 medium containing 0.1% heat-inactivated FBS and then treated with 100 ng/mL recombinant Fgf23 (R&D Systems) or 10 ng/mL LPS for 48 h. Cell numbers were measured with a WST-8 reagent by Cell Count Reagent SF (Nacalai Tesque Inc.) according to the manufacturer’s instructions. Briefly, after treatment with recombinant Fgf23 or LPS for 48 h, WST-8 solution was added to each well, and the cells were incubated for 1 h at 37°C. The absorbance of the cultures was measured at 450 nm. Otherwise, using rubber cell scraper, the cells treated with or without 100 ng/mL Fgf23 for 48 h were detached from the dish. The number of cells was determined by automated cell counter, Countess™ (Invitrogen). In addition, to inhibit the Fgf receptor signaling, adherent cells were incubated in RPMI 1640 medium containing 0.1% heat-inactivated FBS with 10 µM SU5402 (Sigma) for 2 h and then incubated in RPMI 1640 medium containing 0.1% heat-inactivated FBS with or without 100 ng/mL Fgf23 in the presence of 10 µM SU5402 for 48 h. We also pretreated macrophages with 0.5% DMSO and then treated them with or without 100 ng/mL Fgf23 in the presence of 0.5% DMSO. To study the effects of Fgf23 or LPS on cytokine expression, adherent cells were grown for 0, 3, and 24 h in RPMI 1640 containing 0.1% heat-inactivated FBS and either 100 ng/mL mouse recombinant Fgf23 (6His-tagged Tyr25-Val251 [Arg179Gln]; 26.1 kDa, R&D Systems) or 10 ng/mL LPS. Total RNA were extracted from cultured macrophages. IL-6, IL-1β, and TNF-α expression levels were examined by RT-qPCR as described above. In addition, the supernatants of cells treated with or without Fgf23 for 0 and 48 h were collected to determine the TNF-α secretion in medium.
Generation of Bone Marrow (BM)-Derived DCsBM-derived DCs were generated as described previously.9) Briefly, BM cells were flushed out from the femurs and tibias of BALB/c mice with serum-free RPMI-1640 medium through a 2.5-mL syringe. Erythrocytes were lysed using 0.83% NH4Cl solution, and the BM cells (1×106 cells/mL) were cultured in RPMI-1640 medium containing 10% heat-inactivated FBS, 0.03 mg/mL L-glutamine, 100 unit/mL penicillin, and 100 µg/mL streptomycin supplemented with 20 ng/mL GM-CSF and 10 ng/mL IL-4 (Peprotech). After the culture medium containing cytokines was replaced at days 3 and 5. On day 6, nonadherent cells and loosely adherent cells were harvested and used in further experiments. After being cultured in the medium at 37°C for 3h, these cells were cultured in the medium supplied with 100 ng/mL LPS, 0.5 µg/mL Pam3CSK4 (Invivogen), or 1 µM CpG oligodeoxynucleotide (CpG-ODN 1585) (Invivogen) for 3 h to study the induction of Fgf23 expression. To examine the role of NF-κB signaling in Fgf23 expression, 50 mM BAY11-7082 (Merck Millipore) added to the medium 2 h before the addition of LPS.
Western BlotMacrophages (1×106 cells/mL) were stimulated with LPS at 37°C and collected at various time-points by the addition of RIPA buffer (Nacalai Tesque Inc.). Lysate supernatants were resolved by sodium dodecyl sulfate- polyacrylamide gel electrophoresis (SDS-PAGE) and probed with a rabbit anti-β-actin antibody and rabbit anti-inhibitory(I)κBα antibody as the primary antibody and a horseradish peroxidase-conjugated goat anti-rabbit immunoglobulin G (IgG) antibody as the secondary antibody. Western blotting antibodies were purchased from Cell Signaling Technology (Beverly, MA, U.S.A.).
Statistical AnalysisValues are presented as mean±standard error (S.E.). One-way ANOVA with Dunnett’s post-tests was used for analysis of multiple groups. Student’s t-test was used to determine statistical significances to compare two groups. The p values of <0.05 were considered significant.
RESULTS
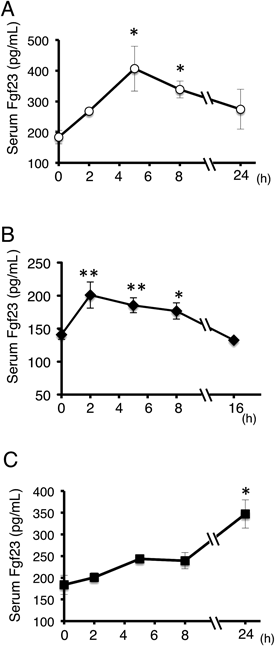
To assess the potential involvement of Fgf23 in immunological responses, serum Fgf23 levels in mice inoculated intraperitoneally with live E. coli were examined. As shown in Fig. 1A, serum Fgf23 levels were increased significantly by the inoculation with 108 CFU E. coli per mouse (Fig. 1A). We also studied the induction of serum Fgf23 levels following treatment with LPS, the major virulence factor of Gram-negative bacteria. LPS stimulates monocytes/macrophages through Toll-like receptor (TLR) 4 activation, promoting a series of signaling events that potentiate the production of inflammatory mediators.10) Serum Fgf23 levels were transiently increased at 2 h following administration of 50 µg/kg LPS to mice, and decreased thereafter (Fig. 1B). Serum Fgf23 levels at 16 h post-LPS administration were comparable to baseline levels observed before the administration. In addition, we examined the levels of serum Fgf23 after the inoculation with live S. aureus, a representative Gram-positive bacterium (Fig. 1C). Intraperitoneal inoculation with 5×108 CFU S. aureus significantly increased serum Fgf23 levels at 24 h post-treatment. These data suggested that Fgf23 levels in blood may increase in response to the immunological stimuli like bacterial infection.
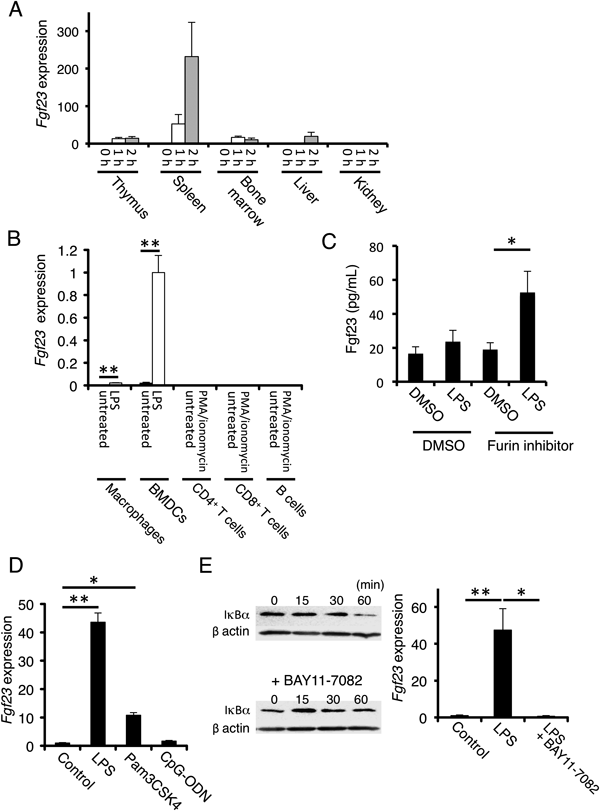
Next, we sought to clarify the organs responsible for the increased blood Fgf23 levels observed following LPS treatment. Fgf23 expression in thymus, spleen, and bone marrow was already induced markedly at 1 h post-administration (Fig. 2A). In liver, Fgf23 expression was also increased at 2 h following administration of LPS. In contrast, Fgf23 expression was almost unchanged in kidney. Notably, Fgf23 was expressed most abundantly in spleen among the tissues tested (Fig. 2A). Therefore, we examined the cells in which Fgf23 was induced in spleen. Our initial hypothesis was that lymphocytes in spleen expressed Fgf23 mRNA in response to the stimuli. PMA and ionomycin are commonly used as stimulants for lymphocytes. Therefore, we determined expression levels of Fgf23 in CD4+ T cells, CD8+ T cells and B cells isolated from spleen with or without the activation by PMA and ionomycin treatment. But Fgf23 was only marginally expressed in these cell types, regardless of treatment (Fig. 2B). In spleen, macrophages and DCs also exist in addition to lymphocytes.11) It is well known that these cells are activated by LPS treatment. Therefore, we studied Fgf23 expression in isolated peritoneal macrophages and in bone marrow-derived DCs (BMDCs). Fgf23 expression was markedly induced in activated BMDCs treated with LPS and was slightly but significantly induced in activated macrophages (Fig. 2B). Using peritoneal macrophages, we also examined the secretion of Fgf23 to cell culture supernatant by ELISA. Unexpectedly, Fgf23 levels in the culture supernatant were unchanged by LPS treatment for 24 h. Fgf23 is cleaved and inactivated by furin/furin-like proprotein convertase cleavage.12) These cleaved Fgf23 could not be detected by commercially available ELISA kit. Therefore, we examined the cleavage of Fgf23 protein using the furin specific inhibitor. In the presence of the inhibitor, the Fgf23 levels were significantly increased by LPS treatment for 24 h (Fig. 2C).
LPS is a ligand for TLR4 that activates macrophages and DCs via binding to them.10) Next we examined Fgf23 expression in BMDCs treated with Pam3CSK4 and CpG-ODN, which are known ligands for TLR2 and TLR9, respectively.10,13,14) Treatment with 0.5 µg/mL Pam3CSK4 significantly increased Fgf23 expression in BMDCs. Fgf23 expression in BMDCs treated with 1 µM CpG-ODN was slightly increased by about 1.7 times but not significantly (Fig. 2D).
TLR signaling exerts multiple effects mainly via the activation of NF-κB.10) Therefore, we examined the role of NF-κB activation on the induction of Fgf23 in BMDCs using BAY11-7082, a pharmacological inhibitor of NF-κB (Merck Millipore). Treatment with LPS for 60 min induces the phosphorylation-dependent degradation of IκBα, the inhibitor of NF-κB, resulting in activation of NF-κB.15) Pretreatment of 50 mM BAY11-7082 almost completely inhibited the degradation of IκBα induced by LPS (Fig. 2E). Although Fgf23 expression in BMDCs was significantly induced by LPS treatment, the pretreatment with BAY11-7082 significantly attenuated Fgf23 induction by LPS (Fig. 2E). These data indicated that the NF-κB activation induced by immunological responses is essential for the induction of Fgf23 expression in DCs.
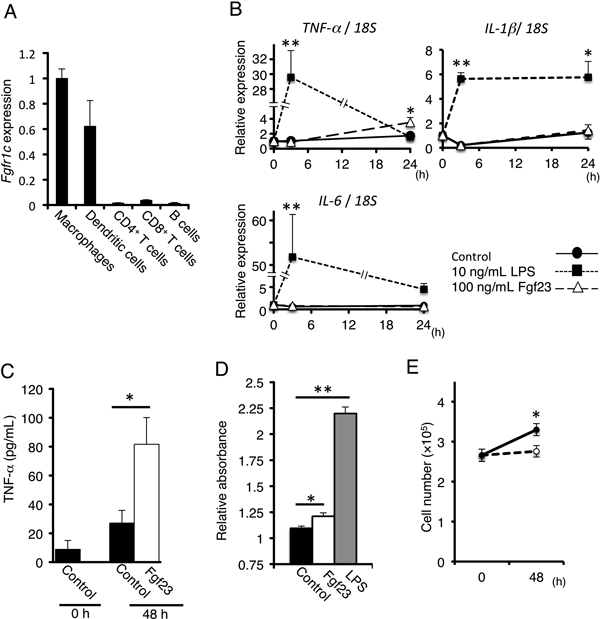
To further study the role of Fgf23 in immune responses, we determined expression levels of Fgfr1c, a potential receptor of Fgf23,16) in a representative panel of immune cells, including macrophages, BMDCs, CD4+ T cells, CD8+ T cells and B cells. Fgfr1c was most abundantly expressed in macrophages, but was also expressed in BMDCs at slightly lower levels. In contrast, Fgfr1c was only marginally detected in CD4+ T cells, CD8+ T cells, and B cells (Fig. 3A). Therefore, we examined the effects of recombinant Fgf23 protein on the expression of cytokines in macrophages. The effects of Fgf23 on cultured cells was often examined by the treatment of 10–100 ng/mL recombinant Fgf23, which is higher than its physiological serum levels.17,18) Therefore, we also treated isolated macrophages with 100 ng/mL recombinant Fgf23. As LPS is known to stimulate cytokine expression in macrophages, we use LPS as a positive control. Both IL-6 and TNF-α expression were induced by LPS treatment for 3 h, and their expression levels were decreased at 24 h post-treatment. The expression of IL-1β was also increased at 3 h post-LPS treatment; however, elevated expression levels were maintained for 24 h. In contrast, treatment of macrophages with 100 ng/mL of recombinant Fgf23 protein for 3 h did not affect the expression of all cytokines studied. Treatment with recombinant Fgf23 for 24 h significantly induced TNF-α expression, the product of which was involved in systemic inflammation (Fig. 3B). TNF-α protein levels in medium were also increased significantly in macrophages treated with 100 ng/mL of recombinant Fgf23 protein for 48 h (Fig. 3C). We also examined the effects of recombinant Fgf23 on the number of macrophages by measuring cell densities using the tetrazolium salt, WST-8 (Fig. 3D). WST-8 is cleaved to soluble formazan dye by the succinate-tetrazolium reductase and is active only in viable cells. The absorbance of formazan dye solution in WST-8 assay was significantly increased by Fgf23 treatment. Although the absorbance of formazan dye solution is believed to be in direct proportion to the number of viable cells, there’s a possibility that the increase in the absorbance is due, not to the increase in the number of macrophages, but to the increased activity of succinate-tetrazolium reductase in each macrophage. Therefore, we also directly confirmed the effects of Fgf23 by counting the cell number of macrophages (Fig. 3E). The number of macrophages was exactly increased by Fgf23 treatment.
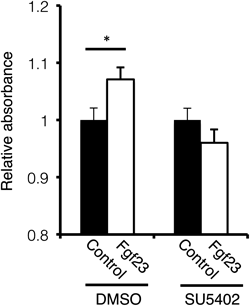
Finally, we examined the role of Fgfr in mediating the effects of Fgf23 in macrophages. The increase in the absorbance of formazan dye solution in WST-8 assay by Fgf23 was inhibited by pretreatment of SU5402 (Sigma), an Fgfr-specific tyrosine kinase inhibitor (Fig. 4). These data suggested that the activation of Fgfr by Fgf23 is essential for the activity of Fgf23 in macrophages.
DISCUSSION
Previous studies have shown that Fgf23 is produced by osteoblasts/osteocytes and regulates the systemic metabolisms of phosphate and vitamin D.3) However, additional physiological roles of Fgf23 almost remain unclear. In the present study, we examined the expression of Fgf23 in immune tissues and cells. Serum Fgf23 level in mice were increased by the inoculation of both E. coli and S. aureus, or intraperitoneal injection of LPS. After LPS administration, Fgf23 was induced in several tissues. Among the tissues tested, Fgf23 was expressed most abundantly in spleen, suggesting that spleen may contribute to the increase in serum Fgf23 levels by the administration of LPS and the inoculation of bacterium in part. In addition, our data about Fgf23 expression using cultured cells suggested that activated macrophages and DCs were potential sources of Fgf23 in spleen. Our in vivo and in vitro data about Fgf23 expression is consistent with the result of cDNA database search by Kato and Kato.7) Regarding the regulatory mechanisms of Fgf23 gene expression, it has been shown that Fgf23 expression is directly induced in osteocytes by ligand-bound vitamin D receptor.19) Recently, Meir et al. reported that an orphan nuclear receptor, Nurr1 also mediates increased Fgf23 expression by parathyroid hormone.20) In this report, we revealed that NF-κB, a common mediator of TLR signaling, is essential for Fgf23 induction at least in cultured DCs. Future studies will be necessary to examine the physiological contribution of DCs and macrophages, and of NF-κB signaling to the serum Fgf23 levels in physiological and pathological conditions, including infectious disease.
Both transcriptional and posttranscriptional mechanisms play important roles in regulating Fgf23 action.12,21) Fgf23 is cleaved between arg179 and ser180 by furin/furin-like proprotein convertase, and this processing abolishes its biologic activity. Only by proinflammatory stimuli, intact FGF23 protein levels in the culture supernatant were unchanged. However, the levels of intact Fgf23 protein were increased by proinflammatory stimuli in the presence of the furin inhibitor. These data indicated that Fgf23 protein induced by proinflammatory stimuli in isolated macrophages was cleaved and inactivated. In addition, considering increased serum Fgf23 levels in mice by the inoculation of bacterium or intraperitoneal injection of LPS, this protease activity in macrophages and DCs in vivo may be lower than that in isolated macrophages and BMDCs in vitro, or may be downregulated in response to immunological stimuli.
We also examined the effects of Fgf23 derived from macrophages and DCs on immune cells. Fgfr1c, which is the potential receptor of Fgf23, was abundantly expressed in macrophages among the diverse immune cell types studied. The expression profile of Fgfr1c prompted us to examine the effects of Fgf23 in macrophages. Although the expression of IL-6 and IL-1β were almost unchanged by treatment with recombinant Fg23, Fgf23 significantly increased the number of macrophages and stimulated TNF-α expression in macrophages. TNF-α induces inflammatory responses as a proinflammatory cytokine.22) Our present data suggested that Fgf23 produced in response to immunological stimuli may induce the inflammatory responses in macrophages.
Although we also examined the expression of α Klotho gene, the product of which functions as a co-receptor for Fgf23, its expression was scarcely detected in all types of immune cells tested, including macrophages (data not shown).3,16) FGF23 was recently shown to induce ventricular hypertrophy in an α Klotho-independent manner.17) In addition, Fgf23 was recently shown to inhibit the secretion of parathyroid hormones from parathyroid glands, both in α Klotho-dependent and -independent manners.23) Our present data suggested that the activation of Fgf receptor by Fgf23 is essential for the activity of Fgf23 in macrophages. Therefore, Fgf23 increases the number of macrophages and induces the expression of TNF-α via the signaling of Fgf receptor in an α Klotho-independent manner.
In summary, we report here that, in addition to osteoblasts/osteocytes, activated macrophages and DCs are also the potential sources of Fgf23. Moreover, recombinant Fgf23 protein increased the number of macrophages and induced TNF-α expression of macrophages. These data suggest that Fgf23 may be involved in the inflammatory process, in addition to the systemic mineral metabolisms.
Conflict of Interest
The authors declare no conflict of interest.
REFERENCES
- 1) Itoh N, Ornitz DM. Fibroblast growth factors: from molecular evolution to roles in development, metabolism and disease. J. Biochem., 149, 121–130 (2011).
- 2) Itoh N. The Fgf families in humans, mice, and zebrafish: their evolutional processes and roles in development, metabolism, and disease. Biol. Pharm. Bull., 30, 1819–1825 (2007).
- 3) Itoh N. Hormone-like (endocrine) Fgfs: their evolutionary history and roles in development, metabolism, and disease. Cell Tissue Res., 342, 1–11 (2010).
- 4) Yamashita T, Yoshioka M, Itoh N. Identification of a novel fibroblast growth factor, FGF-23, preferentially expressed in the ventrolateral thalamic nucleus of the brain. Biochem. Biophys. Res. Commun., 277, 494–498 (2000).
- 5) Shimada T, Hasegawa H, Yamazaki Y, Muto T, Hino R, Takeuchi Y, Fujita T, Nakahara K, Fukumoto S, Yamashita T. FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J. Bone Miner. Res., 19, 429–435 (2004).
- 6) Shimada T, Kakitani M, Yamazaki Y, Hasegawa H, Takeuchi Y, Fujita T, Fukumoto S, Tomizuka K, Yamashita T. Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J. Clin. Invest., 113, 561–568 (2004).
- 7) Katoh Y, Katoh M. Comparative genomics on mammalian Fgf6-Fgf23 locus. Int. J. Mol. Med., 16, 355–358 (2005).
- 8) Masuda Y, Togo T, Mizuno S, Konishi M, Nanba H. Soluble β-glucan from Grifola frondosa induces proliferation and Dectin-1/Syk signaling in resident macrophages via the GM-CSF autocrine pathway. J. Leukoc. Biol., 91, 547–556 (2012).
- 9) Masuda Y, Ito K, Konishi M, Nanba H. A polysaccharide extracted from Grifola frondosa enhances the anti-tumor activity of bone marrow-derived dendritic cell-based immunotherapy against murine colon cancer. Cancer Immunol. Immunother., 59, 1531–1541 (2010).
- 10) Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity, 34, 637–650 (2011).
- 11) Kraal G. Cells in the marginal zone of the spleen. Int. Rev. Cytol., 132, 31–74 (1992).
- 12) Fukumoto S. Post-translational modification of Fibroblast Growth Factor 23. Ther. Apher. Dial., 9, 319–322 (2005).
- 13) Tsolmongyn B, Koide N, Jambalganiin U, Odkhuu E, Naiki Y, Komatsu T, Yoshida T, Yokochi T. A Toll-like receptor 2 ligand, Pam3CSK4, augments interferon-γ-induced nitric oxide production via a physical association between MyD88 and interferon-γ receptor in vascular endothelial cells. Immunology, 140, 352–361 (2013).
- 14) Takeshita F, Leifer CA, Gursel I, Ishii KJ, Takeshita S, Gursel M, Klinman DM. Cutting edge: Role of Toll-like receptor 9 in CpG DNA-induced activation of human cells. J. Immunol., 167, 3555–3558 (2001).
- 15) Hayden MS, Ghosh S. Signaling to NF-κB. Genes Dev., 18, 2195–2224 (2004).
- 16) Goetz R, Nakada Y, Hu MC, Kurosu H, Wang L, Nakatani T, Shi M, Eliseenkova AV, Razzaque MS, Moe OW, Kuro-o M, Mohammadi M. Isolated C-terminal tail of FGF23 alleviates hypophosphatemia by inhibiting FGF23–FGFR–Klotho complex formation. Proc. Natl. Acad. Sci. U.S.A., 107, 407–412 (2010).
- 17) Faul C, Amaral AP, Oskouei B, Hu M-C, Sloan A, Isakova T, Gutiérrez OM, Aguillon-Prada R, Lincoln J, Hare JM, Mundel P, Morales A, Scialla J, Fischer M, Soliman EZ, Chen J, Go AS, Rosas SE, Nessel L, Townsend RR, Feldman HI, St. John Sutton M, Ojo A, Gadegbeku C, Di Marco GS, Reuter S, Kentrup D, Tiemann K, Brand M, Hill JA, Moe OW, Kuro-o M, Kusek JW, Keane MG, Wolf M. FGF23 induces left ventricular hypertrophy. J. Clin. Invest., 121, 4393–4408 (2011).
- 18) Kawai M, Kinoshita S, Kimoto A, Hasegawa Y, Miyagawa K, Yamazaki M, Ohata Y, Ozono K, Michigami T. FGF23 suppresses chondrocyte proliferation in the presence of soluble α-Klotho both in vitro and in vivo. J. Biol. Chem., 288, 2414–2427 (2013).
- 19) Saji F, Shigematsu T, Sakaguchi T, Ohya M, Orita H, Maeda Y, Ooura M, Mima T, Negi S. Fibroblast growth factor 23 production in bone is directly regulated by 1{alpha},25-dihydroxyvitamin D, but not PTH. Am. J. Physiol. Renal Physiol., 299, F1212–F1217 (2010).
- 20) Meir T, Durlacher K, Pan Z, Amir G, Richards WG, Silver J, Naveh-Many T. Parathyroid hormone activates the orphan nuclear receptor Nurr1 to induce FGF23 transcription. Kidney Int., 86, 1106–1115 (2014).
- 21) Bhattacharyya N, Chong WH, Gafni RI, Collins MT. Fibroblast growth factor 23: State of the field and future directions. Trends Endocrinol. Metab., 23, 610–618 (2012).
- 22) Bradley JR. TNF-mediated inflammatory disease. Am. J. Pathol., 214, 149–160 (2008).
- 23) Olauson H, Lindberg K, Amin R, Sato T, Jia T, Goetz R, Mohammadi M, Andersson G, Lanske B, Larsson TE. Parathyroid-specific deletion of Klotho unravels a novel calcineurin-dependent FGF23 signaling pathway that regulates PTH secretion. PLoS Genet., 9, e1003975 (2013).