Regular Articles
Design, Synthesis, and Antitumor Activity of Novel 5-Pyridyl-1,3,4-oxadiazole Derivatives against the Breast Cancer Cell Line MCF-7
2015 Volume 38 Issue 5 Pages 763-773
Details
2015 Volume 38 Issue 5 Pages 763-773
Various 1,3,4-oxadiazole-2-thiol derivatives have considerable potential in the field of antitumor activity. On the basis of the structure of the highly active reported oxadiazole analogues, 36 novel compounds were designed. Their molecular transport properties were predicted using a computer-aided program, and they were then synthesized and tested for anticancer activity against the breast cancer cell line MCF-7. Most of the tested compounds showed excellent to potent cytotoxic activity. Docking studies were carried out to examine the possibilities of the target compounds to become lead compounds in the future after more biological investigations. Compounds 18 and 22 were more active than the reference drug with IC50 values of 0.010 µM and 0.012 µM, respectively, and binding energy scores of −10.32 and −10.25, respectively.
1,3,4-Oxadiazole heterocycles are good bioisosteres of amides and esters, which can contribute substantially in increasing pharmacological activity by participating in hydrogen bonding interactions with the receptors.1–9) Epidermal growth factor receptor (EGFR) is a transmembrane protein tyrosine kinase (PTK) that exists on the cell surface and activated by binding to its specific ligands. Mutations involving EGFR could lead to its constant activation, which resulted in uncontrolled cell division. The up-regulated activity of EGFR correlated with many human tumors, including lung, breast, bladder, prostate and kidney cancers. Therefore, EGFR tyrosine kinase represents an attractive target for the development of novel anti-cancer agents.10) Recently, the EGFR inhibitory activity of certain 2-(benzylsulfanyl)-5-aryl-1,3,4-oxadiazole derivatives (Fig. 1) has been reported.11)

Based on all the aforementioned considerations, thirty six 5-pyridyl-1,3,4-oxadiazolethiols were designed where the carbocyclic anilino moiety was replaced with the more basic heterocyclic pyridyl moiety and benzyl group was replaced with different pharmacophores e.g. aliphatic, hydrazino, hydrazide, heterocyclic or sulphonamide moieties (Fig. 2). The newly synthesized target compounds were evaluated for cytotoxic activity against breast cancer cell line MCF-7, hoping that the new compounds will be of added value to the anticancer library.

The computer-aided prediction of biological activity in relation to the chemical structure of a compound is now a commonly used technique in drug discovery. Parameters like numerical and topological indices are recent outcomes of these techniques, when applied to protein, viral surfaces, ribonucleic acid (RNA) secondary structures and small molecules.12) Fast and reliable estimation of molecular transport properties, particularly intestinal absorption and blood brain barrier penetration, is one of the key factors accelerating the process of drug discovery and development. Traditionally, calculated values of octanol / water partition coefficient (c log P) have been used in this purpose.13) In addition, other parameters have been introduced for absorption prediction, including molecular weight, hydrogen bonding capabilities, and surface properties. A set of rules, known as the “rule of five” introduced by Lipinski has become particularly popular.14–18) After designing the compounds, they were subjected to molecular properties prediction by Molinspiration and MolSoft (MolSoft, 2007) software in order to filter the compounds for synthesis and biological screening and to reduce enormous wastage of expensive chemicals and precious time.
The synthetic strategies and characterization of some novel 1,3,4-oxadiazole-2-thiol derivatives carrying different pharmacophores and heterocyclic rings that are relevant to potential antitumor and cytotoxic activities were described (Charts 1–3). The antitumor activity of the newly synthesized compounds was evaluated according to the protocol of the National Cancer Institute (NCI) in-vitro disease-oriented human cells screening panel assay.
5-Pyridyl-1,3,4-oxadiazole-2-thione/thiol (1) was synthesized by the ring closure reaction of the isonicotinic acid hydrazide with carbon disulfide under basic conditions.19–22) S-Alkylation of compound 1 with either substituted phenacyl bromides, bromochloroethane or ethyl chloroacetate under different basic conditions afforded the key intermediates 2, 3, 6 and 10. Hydrazinolysis of ethanones 2 and 3 gave compounds 4 and 5, respectively (Chart 1).
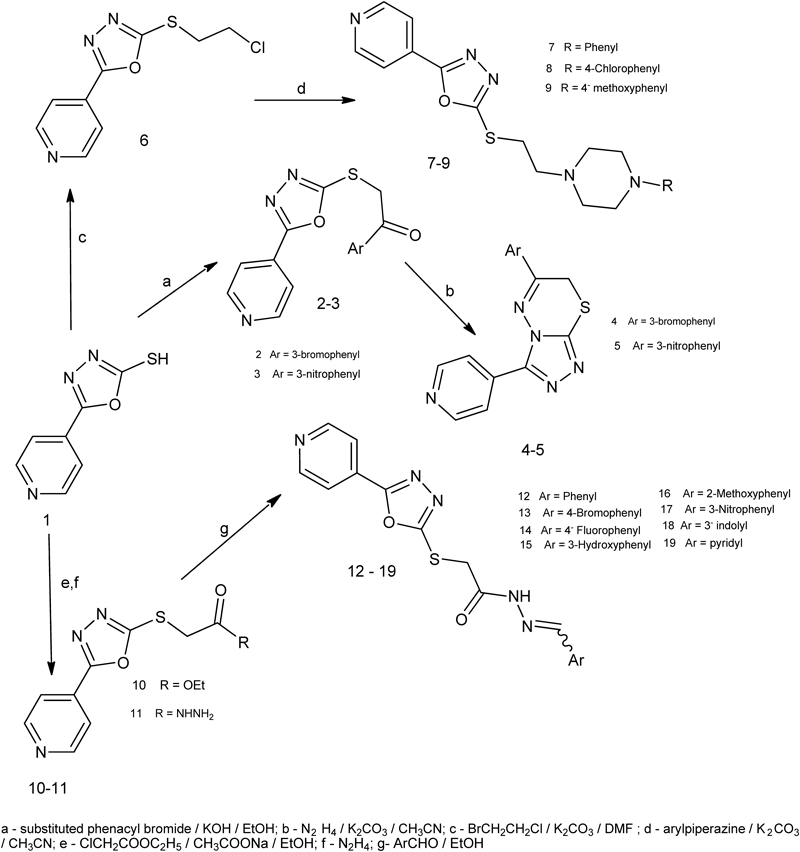
The infrared (IR) spectra of both compounds 4 and 5 showed disappearance of absorption bands corresponding to C=O function. Furthermore, in their proton nuclear magnetic resonance (1H-NMR) spectra, up field shift of singlet signal of SCH2 protons was observed due to ring formation. Nucleophilic substitution of compound 6 with different substituted arylpiperazines afforded the goal compounds 7–9 (Chart 1).
The structure target compounds 7–9 was confirmed by elemental analyses as well as spectroscopic data; IR, 1H-NMR, carbon nuclear magnetic resonance (13C-NMR) and mass spectrum (MS). 1H-NMR spectra showed the emergence of the piperazine ring protons in the range of δ 2.50–2.58 ppm and δ 2.83–3.02 ppm, as well as two triplet signals corresponding to SCH2CH2 protons in the range of δ 2.75–2.79 and δ 3.51–3.54 ppm. 13C-NMR spectrum of the compound 7 revealed signals at δ 48.1 and δ 52.2 ppm corresponding to piperazine ring carbons and signals at δ 30.2 and δ 56.3 ppm attributable to the two aliphatic CH2 groups of the ethyl spacer. Further evidence was substantiated by mass spectrometry. Hydrazinolysis of the ester 10 using hydrazine hydrate at 0°C afforded the back bone intermediate 11 (Chart 1).
It is worth to mention that, higher reaction temperature leads to interconversion of 1,3,4-oxadiazole ring to 4-amino-1,2,4-triazole. During the course of this work, the desired target compounds 12–19 were obtained by the reaction of acetohydrazide derivative 11 with the appropriate aromatic aldehyde in ethanol. Literature survey showed that compounds having arylidine-hydrazide structure may exist as E/Z geometrical isomers about –C=N– and cis/trans amide conformers.23) 1H-NMR of the compounds 12–19 showed the presence of single downfield resonating imine hydrogen signal which accounts for formation of E-isomers exclusively, in DMSO-d6 solution, whereas Z-isomer can be stabilized in less polar solvents by an intramolecular hydrogen bond.24) In the present study, the spectral data were recorded in DMSO-d6, and the signal corresponding to the imine hydrogen was observed in the range of δ 7.95–8.55 ppm.
The investigation of 1H-NMR spectra of 12–19 revealed two sets of signals belonging to each of SCH2, CH=N– and NH groups of cis- and trans-conformers between δ 4.23/δ 7.95/δ 11.78 and δ 4.31/δ 8.55/δ 12.06 ppm. The up field signals of SCH2, N=CH and –NH protons were assigned to cis-conformer of the amide structure and downfield signals of the same group to trans-conformer of the amide structure. 13C-NMR spectrum of compound 15 showed full agreement with the proposed structure. Acylation of the hydrazide 11 with several acyl chlorides in dry acetonitrile afforded the newly synthesized intermediates 20–24 which upon cyclodehydration gave the target compounds 25–29. Moreover reaction of compound 11 with different isothiocyanates yielded compounds 30–32 (Chart 2).

The IR spectra of 20–24 showed an additional C=O functionality. Furthermore, 1H-NMR spectra of compounds 20–24 displayed two D2O exchangeable signals in the range of δ 10.36–10.92 ppm corresponding to two NH protons. Further evidence stemmed from the mass spectra of compounds 20–23, giving base peaks at m/z 105, 139, 133 and 123 respectively, corresponding to acylium cation. IR spectra of compounds 25–29 lacked the presence of an absorption bands due to NH and C=O groups. 1H-NMR spectra showed the disappearance of two signals corresponding to two NH protons. Thiosemicarbazides 30–32 were characterized by the presence of thiourea residue (NH–CS–NH), which can be identified by IR and 1H-NMR spectral data. IR spectra of the title compounds 30–32 exhibited strong absorption band at 1234–1246 cm−1 attributable to C=S. 1H-NMR spectra of 30–32 revealed three D2O exchangeable signals in the range of δ 9.50–10.48 ppm, corresponding to 3NH groups, each showing the integration for one proton.
Sulfonohydrazides 33 and 34 were synthesized via the interaction of compound 11 and the appropriate aryl sulfonyl chloride in dry pyridine (Chart 3). Furthermore, reaction of 11 with carbon disulfide and potassium hydroxide in ethanol at room temperature afforded compound 35. Hydrazinolysis with cyclodesulphorization of compound 35 was supposed to give compound A (Chart 3) but unexpectedly compound 36 was resulted which was structurally confirmed with elemental analysis and spectral data. The reaction of 35 with hydrazine hydrate was carried out at room temperature to avoid interconversion of the oxadiazole to the triazole. Unfortunately, the starting dithiocarbazinate 35 was recovered unchanged. When the reaction was conducted at elevated temperature, compound 36 was obtained in 46% yield.

The structure of the products 33 and 34 was confirmed by elemental analyses, IR, 1H-NMR, 13C-NMR and MS. IR spectra revealed two SO2 str. bands in the range of 1304–1344 and 1158–1161 cm−1. Moreover, 1H-NMR showed appearance of two D2O exchangeable signals in the range of δ 9.93–10.04 and δ 10.41–10.45 ppm, each integrated for one proton, corresponding to two NH protons. In addition, 1H-NMR of 34 displayed a singlet signal at δ 2.29 ppm attributable to CH3 protons. The structure of 35 was confirmed by elemental analysis, IR, 1H-NMR and mass spectrometry. IR spectrum showed the appearance of C=S str. band at 1230 cm−1. Moreover, mass spectrum revealed molecular ion peak at m/z 365. The structure of the new compound 36 was characterized by microanalytical analysis, IR, 1H-NMR and mass spectra. IR spectrum lacked the presence of an absorption band due to C=O. On the other hand, the spectrum revealed a band at 2542 cm−1 corresponding to SH function. 1H-NMR displayed three D2O exchangeable signals at 5.82, 6.28 and 14.14 ppm, due to two NH2 and SH protons, respectively.
Discussion of Computer-Aided PredictionNone of the synthesized compounds violated Lipinski’s parameters, making them potentially promising agents as anticancer agents (Table 1).
From these parameters, it is obvious that, all titled compounds exhibited percentage absorption (% ABS) ranging from 55.23 to 91.12%, among them six compounds are supposed to have very high % ABS (85.23–91.12%). Compounds 6, 4, 7–9 and 2 showed greater % ABS than that of the reference drug erlotinib (83.21%). All the newly synthesized compounds showed good membrane permeability since they all exhibited c Log P≤5. Compounds 7 and 9 were similar to that of erlotinib. The number of rotatable bonds (NROTB) ranged from 2–9 which will facilitate the conformational changes required for these compounds to fit inside the receptors.
| Compound | % ABS | TPSA | NROTB | HBA | HBD | c Log P | MW | Lipinski’s violations |
|---|---|---|---|---|---|---|---|---|
| Rule | — | — | ≤10 | ≤10 | ≤5 | ≤5 | ≤500 | ≤1 |
| Erlotinib | 83.21 | 74.74 | 10 | 7 | 1 | 2.81 | 393.44 | 0 |
| 2 | 85.23 | 68.88 | 5 | 5 | 0 | 3.17 | 376.23 | 0 |
| 3 | 69.42 | 114.71 | 6 | 8 | 0 | 2.32 | 342.33 | 0 |
| 4 | 89.69 | 55.97 | 2 | 5 | 0 | 3.35 | 372.25 | 0 |
| 5 | 73.88 | 101.79 | 3 | 8 | 0 | 2.50 | 338.35 | 0 |
| 6 | 91.12 | 51.81 | 4 | 4 | 0 | 1.84 | 241.70 | 0 |
| 7 | 88.88 | 58.29 | 6 | 6 | 0 | 2.85 | 367.47 | 0 |
| 8 | 88.88 | 58.29 | 6 | 6 | 0 | 3.53 | 401.92 | 0 |
| 9 | 85.70 | 67.52 | 7 | 7 | 0 | 2.91 | 397.50 | 0 |
| 12 | 76.82 | 93.27 | 6 | 7 | 1 | 2.31 | 339.38 | 0 |
| 13 | 76.82 | 93.27 | 6 | 7 | 1 | 3.12 | 418.27 | 0 |
| 14 | 76.82 | 93.27 | 6 | 7 | 1 | 2.47 | 357.37 | 0 |
| 15 | 69.84 | 113.50 | 6 | 8 | 2 | 1.81 | 355.37 | 0 |
| 16 | 73.63 | 102.51 | 7 | 8 | 1 | 2.32 | 369.40 | 0 |
| 17 | 61.01 | 139.10 | 7 | 10 | 1 | 2.22 | 384.37 | 0 |
| 18 | 72.37 | 106.17 | 6 | 8 | 1 | 1.02 | 340.36 | 0 |
| 19 | 71.37 | 109.06 | 6 | 8 | 2 | 2.46 | 378.41 | 0 |
| 20 | 71.04 | 110.01 | 6 | 8 | 2 | 1.13 | 355.37 | 0 |
| 21 | 71.04 | 110.01 | 6 | 8 | 2 | 1.81 | 389.82 | 0 |
| 22 | 71.04 | 110.01 | 6 | 8 | 2 | 1.25 | 373.36 | 0 |
| 23 | 55.23 | 155.83 | 7 | 11 | 2 | 1.09 | 400.37 | 1 |
| 24 | 71.04 | 110.01 | 7 | 8 | 2 | 2.05 | 383.43 | 0 |
| 25 | 77.69 | 90.74 | 5 | 7 | 0 | 2.25 | 337.36 | 0 |
| 26 | 77.69 | 90.74 | 5 | 7 | 0 | 2.93 | 371.80 | 0 |
| 27 | 77.69 | 90.74 | 5 | 7 | 0 | 2.37 | 355.35 | 0 |
| 28 | 61.88 | 136.56 | 6 | 10 | 0 | 2.21 | 382.36 | 0 |
| 29 | 77.69 | 90.74 | 6 | 7 | 0 | 3.17 | 365.41 | 0 |
| 30 | 72.78 | 104.96 | 8 | 8 | 3 | 1.35 | 386.46 | 0 |
| 31 | 72.78 | 104.96 | 8 | 8 | 3 | 2.03 | 420.90 | 0 |
| 32 | 69.60 | 114.20 | 9 | 9 | 3 | 1.41 | 416.48 | 0 |
| 33 | 65.18 | 127.00 | 7 | 9 | 2 | 1.01 | 391.43 | 0 |
| 34 | 65.15 | 127.08 | 7 | 9 | 2 | 1.46 | 405.46 | 0 |
| 36 | 65.40 | 126.37 | 4 | 9 | 4 | −0.09 | 321.39 | 0 |
% ABS=% Absorption. TPSA=Topological polar surface area. NROTB=Number of rotatable bonds. HBA=Hydrogen bond acceptors. HBD=Hydrogen bond donors. Log P=Octanol/water partition coefficient. MW=Molecular weight.
All the newly synthesized compounds were tested for anticancer activity against breast carcinoma cell line MCF-7 using sulforhodamine B (SRB) assay and using erlotinib as a reference drug.25) The concentration required for 50% inhibition of cell viability (IC50) values were calculated in micromole (µM) and compared with that of erlotinib (IC50=0.020 µM). The results are summarized in Table 2.
| Compd. No. | IC50 in µM/mL | Compd. No. | IC50 in µM/mL | Compd. No. | IC50 in µM/mL |
|---|---|---|---|---|---|
| 2 | 0.063 | 3 | 0.043 | 4 | — |
| 5 | 0.059 | 6 | 0.081 | 7 | 0.063 |
| 8 | 0.045 | 9 | 0.080 | 12 | 0.128 |
| 13 | 0.043 | 14 | 0.105 | 15 | 0.070 |
| 16 | 0.061 | 18 | 0.010 | 19 | — |
| 20 | 0.061 | 21 | 0.050 | 22 | 0.012 |
| 23 | 0.036 | 24 | 0.111 | 28 | 0.056 |
| 29 | 0.037 | 30 | 0.122 | 31 | 0.046 |
| 32 | — | 33 | — | 34 | 0.075 |
| 36 | 0.063 | Erlotinib | 0.020 |
All tested compounds exhibited significant anticancer activity against breast carcinoma cell line MCF-7 with IC50 values range (0.010 and 0.081) µM. Compounds 18 and 22 showed more potent cytotoxic activity than the reference drug Erlotinib with IC50=0.010 and 0.012 µM, respectively.
Moreover, 23 and 29 showed good cytotoxic activity with IC50 0.036 µM and 0.037 µM, respectively. Compounds 3, 8, 13 and 31 showed nearly half the potency of the reference drug with IC50 values 0.043, 0.045, 0.043 and 0.046 µM. Compounds 2, 5, 7, 15, 16, 20, 21, 28 and 36 were nearly one third the potency of the reference drug with IC50 ranges from 0.050 to 0.070 µM. Compounds 34, 6 and 9 showed moderate cytotoxic activity while compounds 12, 14, 24 and 30 were nearly one tenth the antitumor activity of the reference drug. Compounds 4, 19, 32 and 33 revealed no cytotoxic activity.
Discussion of Docking StudiesAmino acid interactions and hydrogen bond lengths of the biologically active compounds with the active site of EGFR (PDB ID: 1M17) were summarized and compared to that of erlotinib (Table 3). In the molecular modeling study, the biologically active compounds were docked into the binding site of EGFR-TK using molecular operating environment (MOE) program.
| Compound No. | No. of H-bonds | Amino acid residues forming H-bonds (H-bond length in Å) | Binding energy score* |
|---|---|---|---|
| Erlotinib | 2 | Meth769 (2.7 Å) and Thr766 (2.95 Å). | −10.88 |
| 2 | 2 | Meth769 (3 Å) and Lys721 (arene–cation interaction). | −10.72 |
| 3 | 2 | Meth769 (3 Å) and Lys721 (arene–cation interaction). | −11.42 |
| 5 | 2 | Thr766 (2.95 Å) and Lys721 (2.5 Å). | −8.60 |
| 7 | 2 | Lys 692 (2.7 Å) and Lys 721 (arene–cation interaction). | −10.33 |
| 8 | 2 | Thr766 (3 Å) and Lys721 (2.7 Å). | −8.91 |
| 13 | 2 | Meth769 (2.9 Å) and Lys692 (2.7 Å). | −10.32 |
| 15 | 3 | Meth769 (3 Å), Thr766 (2.7 Å) and Lys692 (2.8 Å). | −10.64 |
| 16 | 4 | Meth769 (3 Å), Thr766 (3 Å), (3.25 Å) and Lys692 (2.9 Å). | −10.61 |
| 18 | 2 | Meth769 (2.9 Å) and Lys721 (arene–cation interaction). | −10.32 |
| 20 | 1 | Meth769 (3 Å). | −9.36 |
| 21 | 1 | Meth769 (3 Å). | −10.27 |
| 22 | 2 | Meth769 (2.9 Å) and Lys692 (2.7 Å). | −10.25 |
| 23 | 2 | Meth769 (3 Å) and Lys692 (2.7 Å). | −9.98 |
| 25 | 1 | Thr766 (3 Å). | −8.79 |
| 28 | 1 | Meth769 (3 Å). | −10.99 |
| 29 | 1 | Thr766 (2.95 Å). | −9.79 |
| 31 | 1 | Meth769 (2.7 Å). | −11.00 |
| 34 | 2 | Meth769 (2.9 Å) and Lys692 (2.6 Å). | −10.90 |
| 36 | 1 | Meth769 (2.7 Å). | −9.69 |
* Binding energy score (kcal/mol): energy of interaction of the ligand in the active site. The higher the score in negative terms, the better the binding affinity.
Docking studies revealed that, all the docked compounds showed interaction with EGFR at the ATP binding site with an estimated binding free energy scores ranging from −8.60 to −11.42 kcal/mol. The hydrogen bonding with Met769, which is considered a critical amino acid residue in the active site EGFR-TK, helps to fix the adenine base of adenosine triphosphate (ATP) in the binding pocket and considered crucial for the activity of the anticancer agents.
In this study, H-bond interaction with Met769 was observed in most of the docked compounds. The most potent compound in cytotoxicity assay 18 showed binding to Met769 through H-bond interaction (distance=2.9 Å), as well as arene–cation interaction between the indole ring and Lys721. The latter interaction afforded stabilization of the ligand/receptor complex, and this multi-bond pattern could keep the compound in a favorable conformation (Fig. 3). Furthermore, the indole ring is deeply oriented in the hydrophobic pocket at the back of ATP binding site while pyridine ring is extended to the solvent exposed region.

Compounds 22 displayed binding to Met769 and Lys692 through two H-bonds. Ethyl substituent in 22 is directed toward the back of ATP binding site, making predominant hydrophobic interaction with the active site of EGFR. Additional binding energy provided by hydrophobic interaction of the indole ring (Fig. 4) and ethyl substituent in 18 and 22 may be considered a reason for the enhanced binding affinity and cytotoxic activity of these compounds.

We have designed and synthesized a series of 1,3,4-oxadiazolethioethers to investigate the effect of the replacement of the anilino moiety with the heterocyclic pyridyl moiety, as well as the replacement of the benzyl group at its 2 position with lipophilic, polar and basic pharmacophores against breast cancer cell line MCF-7. The computer-aided prediction of the biological activity in relation to the chemical structure of the newly synthesized compounds showed that most of these compounds have greater or almost equal % ABS to the reference drug. The antitumor activity revealed that compounds 18 and 22 are almost double the potency of the reference drug, this could be attributed to their chemical structure that allows them to have high topological polar surface area (TPSA) that allow good passive molecular transport through membranes. Also they have more hydrogen bond acceptors (HBA) (8) than the reference drug and they both have (6) NROTB that can form a large number of conformational changes that allow them to fit into the receptor. While compounds 23 and 29 are almost as potent as the reference drug, the other compounds showed moderate anticancer activity. This may be attributed to their high ability to high %ABS. From the Docking studies’ results we may conclude that these biologically active compounds with future further investigations could form a potential lead compounds for enriching the anticancer libraries since they interacted smoothly with EGFR at the ATP binding site.
Melting points were determined on Stuart apparatus and the values given are uncorrected. IR spectra were determined on Shimadzu IR 8400 s spectrophotometer (KBr, cm−1). 1H-NMR spectra were carried out using Mercury 300-BB 300 MHz using TMS as internal standard. Chemical shift values were recorded in ppm on δ scale, Microanalytical center, Cairo University, Egypt. Gemini 300-BB 300 MHz using TMS as internal standard. Chemical shift values were recorded in ppm on δ scale, Main Laboratory of Chemical War Department, Ministry of Defense, Egypt. Joel (eca) 500 MHz spectrophotometer using tetramethyl silane (TMS) as internal standard. Chemical shift values were recorded in ppm on δ scale. National Research Centre, Egypt. 13C-NMR spectra were carried out using a Mercury 300-BB 75 MHz spectrophotometer using TMS as internal standard. Chemical shift values were recorded in ppm on δ scale, Microanalytical center, Cairo University, Egypt. Mass spectra were recorded on Hewlett Packard 5988 spectrometer, Microanalytical Center, Cairo University, Egypt. Elemental analyses were carried out at the Regional Center for Mycology and Biotechnology, Faculty of Pharmacy, Al Azhar University, Egypt. Progress of the reactions was monitored using thin-layer chromatography (TLC) sheets precoated with ultraviolet (UV) fluorescent silica gel Merck 60F 254. The solvent system was benzene, chloroform and methanol (5 : 9 : 1) and spots were visualized using UV lamp. The docking was performed using Molecular Operating Environment (MOE 2008.10). Evaluation of the cytotoxic activity was performed at the Egyptian National Cancer Institute. Compounds 5-(pyridin-4-yl)-1,3,4-oxadiazole-2-thione/thiol (1) was synthesized according to reported method.19) Ethyl 2-[(5-(pyridin-4-yl)-1,3,4-oxadiazol-2-ylsulphanyl]acetate (10).26) 2-[5-(Pyridin-4-yl)-1,3,4-oxadiazol-2-ylsulphanyl]acetohydrazide (11).27)
General Procedure for the Preparation of 1-Aryl-2-[5-(pyridin-4-yl)-1,3,4-oxadiazol-2-ylsulphanyl]ethanones (2, 3)A solution of the appropriate phenacyl bromide (0.003 mol) in ethanol (80%, 10 mL) was added to a solution of an equimolar mixture of compound 1 (0.53 g, 0.003 mol) and potassium hydroxide (0.17 g, 0.003 mol) in ethanol (80%, 20 mL). The reaction mixture was stirred for 18 h at room temperature, after which; water (25 mL) was added and the resulting solid product was filtered then crystallized from ethanol to give compounds 2 and 3.
1-Bromophenyl-2-[5-(pyridin-4-yl)-1,3,4-oxadiazol-2-ylsulphanyl]ethanone (2)Yield, 76%; mp 175–176°C; 1H-NMR (DMSO-d6, 500 MHz) δ: 5.18 (2H, s), 7.53 (1H, t, J=7.6, 7.6 Hz), 7.85 (2H, d, J=6 Hz), 7.89 (1H, d, J=7.6 Hz), 8.03 (1H, d, J=7.6 Hz), 8.19 (1H, s), 8.78 (2H, d, J=6 Hz). IR (KBr) cm−1: 3012, 2981, 2908, 1678, 1608. Electrospray ionization (ESI)-MS m/z: 377.20 (M+2+·), 375.20 (M+·), 183.05, 185.05, 106.15, 78.10, 51.05. Anal. Calcd for C15H10BrN3O2S: C, 47.89; H, 2.68; N, 11.17. Found: C, 47.96; H, 2.72; N, 11.43.
1-Nitrophenyl-2-[5-(pyridin-4-yl)-1,3,4-oxadiazol-2-ylsulphanyl]ethanone (3)Yield, 48%; mp 182–183°C; 1H-NMR (DMSO-d6, 500 MHz) δ: 5.29 (2H, s), 7.80–7.95 (3H m, 2H), 8.49 (2H, t, J=7.6, 7.6 Hz), 8.76 (1H, s), 8.81 (2H, d, J=6 Hz). 13C-NMR (DMSO-d6, 500 MHz) δ: 40.6, 119.9, 122.8, 128.9, 129.9, 130.7, 134.6, 136.1, 147.9, 150.8, 163.7, 164.5, 191.3. IR (KBr) cm−1: 3086, 3039, 2916, 2850, 1681, 1612. ESI-MS m/z: 342.25 (M+·), 150.10, 106.15, 78.10, 51.05. Anal. Calcd for C15H10N4O4S: C, 52.63; H, 2.94; N, 16.37. Found: C, 52.74; H, 2.98; N, 16.58.
General Procedure for the Preparation of 6-Aryl-3-(pyridin-4-yl)-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazines (4, 5)To a solution of compound 2 or 3 (0.001 mol) in glacial acetic acid (5 mL), hydrazine hydrate (99%, 0.10 g, 0.002 mol) was added. The reaction mixture was heated under reflux for 8 h then poured onto ice-cold water. The resulted precipitate was filtered, dried then crystallized from ethanol to give 4 and 5.
6-Bromophenyl-3-(pyridin-4-yl)-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazine (4)Yield, 58%; mp 210–211°C; 1H-NMR (DMSO-d6, 300 MHz) δ: 4.46 (2H, s), 7.57 (1H, t, J=6.9, 6.9 Hz), 7.84 (1H, d, J=6.9 Hz), 7.99 (2H, d, J=6 Hz), 8.04 (1H, d, J=6.9 Hz), 8.20 (1H, s), 8.80 (2H, d, J=6 Hz). 13C-NMR (DMSO-d6, 300 MHz) δ: 22.9, 121.4, 122.3, 126.6, 130.3, 131.2, 132.8, 134.7, 135.5, 145.0, 150.3, 155.2, 160.0. IR (KBr) cm−1: 3050, 2997, 1600. ESI-MS m/z: 373.10 (M+2+·), 372.10 (M+H+·), 371.10 (M+·), 78.05, 51.05. Anal. Calcd for C15H10BrN5S: C, 48.40; H, 2.71; N, 18.81. Found: C, 48.47; H, 2.81; N, 19.12.
6-Nitrophenyl-3-(pyridin-4-yl)-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazine (5)Yield, 51%; mp 220–221°C; 1H-NMR (DMSO-d6, 300 MHz) δ: 4.55 (2H, s), 7.89 (1H, t, J=7.8, 7.8 Hz), 7.98 (2H, d, J=4.8 Hz), 8.45 (2H, d, J=7.8 Hz), 8.79 (2H, d, J=4.8 Hz), 8.80 (1H, s). IR (KBr) cm−1: 3086, 2912, 2870, 1604. ESI-MS m/z: 338.10 (M+·), 217.00, 78.00, 51.05. Anal. Calcd for C15H10N6O2S: C, 53.25; H, 2.98; N, 24.84. Found: C, 53.47; H, 3.10; N, 25.19%.
2-(2-Chloroethylsulphanyl)-5-(pyridin-4-yl)-1,3,4-oxadiazole (6)A mixture of 1 (0.89 g, 0.005 mol), 1-bromo-2-chloroethane (3.58 g, 0.025 mol) and anhydrous potassium carbonate (3.45 g, 0.025 mol) in dry N,N-dimethylformamide (DMF) (15 mL) was stirred 12 h at room temperature then poured onto ice-cold water (20 mL). The precipitate so formed was filtered, dried and crystallized from benzene in Yield, 67%; mp 96–97°C; 1H-NMR (DMSO-d6, 500 MHz) δ: 3.71 (2H, t, J=6.9, 6.9 Hz), 4.01 (2H, t, J=6.9, 6.9 Hz), 7.91 (2H, d, J=4.5 Hz), 8.82 (2H, d, J=4.5 Hz). IR (KBr) cm−1: 3039, 2997, 1608. ESI-MS m/z: 243.35 (M+2+·), 241.45 (M+·), 179.35, 146.65, 79.35, 51.35. Anal. Calcd for C9H8ClN3OS: Calcd, C, 44.72; H, 3.34; N, 17.39, Found, C, 44.79; H, 3.38; N, 17.43.
General Procedure for the Preparation of 2-[2-(4-Aryl-piperazin-1-yl)ethylsulphanyl]-5-(pyridin-4-yl)-1,3,4-oxadiazoles (7–9)To a solution of 6 (0.24 g, 0.001 mol) in dry acetonitrile (20 mL), anhydrous potassium carbonate (0.69 g, 0.005 mol) and few specs of potassium iodide (0.05 g) were added. The reaction mixture was heated under reflux for 0.5 h, then an appropriate phenylpiperazine (0.003 mol) was added, and the heating was continued for 20 h. After cooling, the reaction mixture was poured onto ice-cold water with continuous stirring. The precipitate formed was filtered, washed with cold ether, dried then crystallized from isopropanol to give compounds 7–9.
2-[2-(4-Phenyl-piperazin-1-yl)ethylsulphanyl]-5-(pyridin-4-yl)-1,3,4-oxadiazole (7)Yield, 50%; mp 112–113°C. 1H-NMR (DMSO-d6, 500 MHz) δ: 2.50–2.56 (4H, m), 2.75 (2H, t, J=6.1, 6.1 Hz), 2.90–3.02 (4H, m), 3.51 (2H, t, J=6.1, 6.1 Hz), 6.72 (1H, t, J=8.4, 8.4 Hz), 6.85 (2H, d, J=8.4 Hz), 7.15 (2H, t, J=8.4, 8.4 Hz), 7.86 (2H, d, J=5.4 Hz), 8.77 (2H, d, J=5.4 Hz). 13C-NMR (DMSO-d6): 30.2, 48.1, 52.2, 56.3, 115.3, 118.8, 119.8, 128.8, 130.0, 130.1, 150.8, 163.3, 165.7, IR (KBr) cm−1:3035, 2951, 2819, 1600. ESI-MS m/z: 367.00 (M+·), 189.05, 175.05, 78.00. Anal. Calcd for C19H21N5OS: C, 62.10; H, 5.76; N, 19.06. Found: C, 62.17; H, 5.82; N, 19.12.
2-[2-(4-(4-Chlorophenyl)-piperazin-1-yl)ethylsulphanyl]-5-(pyridin-4-yl)-1,3,4-oxadiazole (8)Yield, 60%; mp 120–121°C; 1H-NMR (DMSO-d6, 300 MHz) δ: 2.50–2.54 (4H, m), 2.75 (2H, t, J=6.1, 6.1 Hz), 2.90–3.02 (4H, m), 3.51 (2H, t, J=6.1, 6.1 Hz), 6.87 (2H, d, J=9.2 Hz), 7.17 (2H, d, J=9.2 Hz), 7.86 (2H, d, J=5.4 Hz), 8.77 (2H, d, J=5.4 Hz). IR (KBr) cm−1: 3039, 2924, 2816, 1593. ESI-MS m/z: 403.20 (M+2+·), 401.20 (M+·), 225.10, 223.10, 211.10, 209.10, 78.05. Anal. Calcd for C19H20ClN5OS: C, 56.78; H, 5.02; N, 17.43. Found: C, 56.74; H, 5.12; N, 17.67.
2-[2-(4-(4-Methoxyphenyl)-piperazin-1-yl)ethylsulphanyl]-5-(pyridin-4-yl)-1,3,4-oxadiazole (9)Yield, 49%; mp 125–126°C; 1H-NMR (DMSO-d6, 300 MHz) δ: 2.50–2.58 (4H, m), 2.79 (2H, t, J=6.3, 6.3 Hz), 2.83–2.93 (4H, m), 3.54 (2H, t, J=6.3, 6.3 Hz), 3.67 (3H, s), 6.75–6.95 (4H, m), 7.89 (2H, d, J=5.8 Hz), 8.80 (2H, d, J=5.8 Hz). IR (KBr) cm−1: 3050, 2816, 1600. ESI-MS m/z: 397.20 (M+·), 219.15, 205.15, 78.05, 70.10. Anal. Calcd for C20H23N5O2S: C, 60.43; H, 5.83; N, 17.62. Found: C, 60.52; H, 5.94; N, 17.93.
General Procedure for the Preparation of (Z/E) N′-Arylidene-2-[5-(pyridin-4-yl)-1,3,4-oxadiazol-2-ylsulphanyl]acetohydrazides (12–19)A solution of equimolar amounts of 11 (0.25g, 0.001 mol) and the appropriate aromatic aldehyde (0.001 mol) in absolute ethanol (15 mL) was heated under reflux for 3 h. After cooling, the formed solid product was filtered, dried then crystallized from acetic acid to give 12–19.
(Z/E) N′-Benzilidene-2-[5-(pyridin-4-yl)-1,3,4-oxadiazol-2-ylsulphanyl]acetohydrazide (12)Yield, 56%; mp 201–202°C; 1H-NMR (DMSO-d6, 500 MHz) δ: 4.27, 4.69 (2H, 2s), 7.42–8.81 (9H, m), 8.05, 8.22 (1H, 2s), 11.84 (1H, s). IR (KBr) cm−1: 3170, 3074, 2947, 2858, 1681, 1616. ESI-MS m/z: 339.00, 179.00, 146.10, 106.05, 78.05, 51.00. Anal. Calcd for C16H13N5O2S: C, 56.63; H, 3.86; N, 20.64. Found: C, 56.74; H, 3.91; N, 20.92%.
N′-[(Z/E)-(4-Bromophenyl)methylidene]-2-{[5-(pyridin-4-yl)-1,3,4-oxadiazol-2-yl]sulfanyl}acetohydrazide (13)Yield, 65%; mp 196–197°C; 1H-NMR (DMSO-d6, 300 MHz) δ: 4.27, 4.70 (2H, 2s), 7.64–8.81 (8H, m), 8.01, 8.18 (1H, 2s), 11.89, 11.91 (1H, 2s). IR (KBr) cm−1: 3205, 3051, 2935, 2816, 1689, 1608. ESI-MS m/z: 417.00 (M+·), 77.00, 51.00. Anal. Calcd for C16H12BrN5O2S: C, 45.94; H, 2.89; N, 16.74. Found: C, 45.95; H, 2.89; N, 16.98%.
N′-[(Z/E)-(4-Fluorophenyl)methylidene]-2-{[5-(pyridin-4-yl)-1,3,4-oxadiazol-2-yl]sulfanyl}acetohydrazide (14)Yield, 93%; mp 200–201°C; 1H-NMR (DMSO-d6, 300 MHz) δ: 4.27, 4.70 (2H, s), 7.25–8.80 (8H, m), 8.03, 8.21 (1H, 2s), 11.84, 11.86 (1H, 2s). IR (KBr) cm−1: 3421, 3055, 2939, 2827, 1690, 1612. ESI-MS m/z: 357.00 (M+·), 179.00, 96.00, 70.00. Anal. Calcd for C16H12FN5O2S: C, 53.77; H, 3.38; N, 19.60. Found: C, 53.91; H, 3.42; N, 19.79%.
N′-[(Z/E)-(3-Hydroxyphenyl)methylidene]-2-{[5-(pyridin-4-yl)-1,3,4-oxadiazol-2-yl]sulfanyl}acetohydrazide (15)Yield, 59%; mp 220–221°C; 1H-NMR (DMSO-d6, 500 MHz) δ: 4.26, 4.69 (2H, 2s), 6.80–8.81 (8H, m), 7.95, 8.11 (1H, 2s), 9.64 (1H, s), 11.78, 11.82 (1H, 2s). 13C-NMR (DMSO-d6): 34.87, 112.61, 117.37, 118.8, 119.9, 121.3, 129.8, 135.1, 144.4, 150.2, 157.7, 163.5, 164.9, 167.7. IR (KBr) cm−1: 3400 (OH), 3186, 3070, 2978, 2870, 1670, 1612. ESI-MS m/z: 355.00 (M+·), 106.05, 78.00, 50.95. Anal. Calcd for C16H13N5O3S: C, 54.08; H, 3.69; N, 19.71. Found: C, 54.12; H, 3.58; N, 19.84%.
N′-[(Z/E)-(2-Methoxyphenyl)methylidene]-2-{[5-(pyridin-4-yl)-1,3,4-oxadiazol-2-yl]sulfanyl}acetohydrazide (16)Yield, 82% (150 mg); mp 180–181°C; 1H-NMR (DMSO-d6, 300 MHz) δ: 3.85 (3H, s), 4.23, 4.68 (2H, 2s), 6.96–8.79 (8H, m), 8.37, 8.55 (1H, 2s), 11.78, 11.87 (1H, 2s). IR (KBr) cm−1: 3201, 3066, 2935, 2839, 1685, 1608. ESI-MS m/z: 369.00 (M+·), 107.00, 106.05, 78.00, 51.00. Anal. Calcd for C17H15N5O3S: C, 55.27; H, 4.09; N, 18.96. Found: C, 55.33; H, 4.19; N, 19.32%.
N′-[(Z/E)-(4-Nitrophenyl)methylidene]-2-{[5-(pyridin-4-yl)-1,3,4-oxadiazol-2-yl]sulfanyl}acetohydrazide (17)Yield, 66%; mp 216–217°C; 1H-NMR (DMSO-d6, 500 MHz) δ: 4.31, 4.76 (2H, 2s), 7.70–8.80 (8H, m), 8.35, 8.53 (1H, 2s), 12.06, 12.13 (1H, 2s). IR (KBr) cm−1: 3421, 3074, 2939, 2823, 1685, 1608. ESI-MS m/z: 384.00 (M+·), 179.00, 106.00, 78.00, 51.00. Anal. Calcd for C16H12N6O4S: C, 50.00; H, 3.15; N, 21.86. Found: C, 50.11; H, 3.23; N, 22.14%.
N′-[(Z/E)-(3-Indolyl)methylidene]-2-{[5-(pyridin-4-yl)-1,3,4-oxadiazol-2-yl]sulfanyl}acetohydrazide (18)Yield, 80%; mp 242–243°C. 1H-NMR (DMSO-d6, 500 MHz) δ: 4.25, 4.77 (2H, 2s), 7.11–8.80 (9H, m), 8.22, 8.37 (1H, 2s), 11.50, 11.59 (2H, 2s). IR (KBr) cm−1: 3263, 3104, 2863, 1671, 1617. ESI-MS m/z: 377.95 (M+·), 142.05, 129.05, 116.05, 78.00, 50.95. Anal. Calcd for C18H14N6O2S: C, 57.13; H, 3.73; N, 22.21. Found: C, 57.17; H, 3.79; N, 22.28.
N′-[(Z/E)-(4-Pyridyl)methylidene]-2-{[5-(pyridin-4-yl)-1,3,4-oxadiazol-2-yl]sulfanyl}acetohydrazide (19)Yield, 59% (150 mg); mp 206–207°C; 1H-NMR (DMSO-d6, 500 MHz) δ: 4.29, 4.71 (2H, 2s), 7.64 (2H, d, J=5.4 Hz), 7.88 (2H, d, J=5.7 Hz), 8.03, 8.23 (1H, 2s), 8.62 (2H, d, J=5.4 Hz), 8.79 (2H, d, J=5.7 Hz), 12.02 (1H, s). IR (KBr) cm−1: 3441, 3078, 2924, 2816, 1681, 1597. ESI-MS m/z: 340.10 (M+·), 145.05. Anal. Calcd for C15H12N6O2S: C, 52.93; H, 3.55; N, 24.69. Found: C, 53.04; H, 3.59; N, 24.77.
General Procedure for the Preparation of N′-{2-[5-(Pyridin-4-yl)-1,3,4-oxadiazol-2-ylsulphanyl]acetyl}-Substituted Benzohydrazides (20–24)A mixture of equimolar amounts of 11 (0.25g, 0.001 mol) and the appropriate benzoyl chloride (0.001 mol) in dry acetonitrile (25 mL) was heated under reflux for 4 h, and then cooled. After cooling, the resulted solid was filtered, dried then crystallized from ethanol to give compounds 20–24.
N′-{2-[5-(Pyridin-4-yl)-1,3,4-oxadiazol-2-ylsulphanyl]acetyl}benzohydrazide (20)Yield, 92%; mp 214–215°C; 1H-NMR (DMSO-d6, 500 MHz) δ: 4.26 (2H, s), 7.45–8.83 (9H, m), 10.36 (2H, br s). IR (KBr) cm−1: 3178–3109, 3008, 2958, 2873, 1693, 1643, 1627. ESI-MS m/z: 354.95 (M+·), 105.00, 77.00. Anal. Calcd for C16H13N5O3S: C, 54.08; H, 3.69; N, 19.71. Found: C, 54.11; H, 3.74; N, 19.83%.
4-Chloro-N′-({[5-(pyridin-4-yl)-1,3,4-oxadiazol-2-yl]sulfanyl}acetyl)benzohydrazide (21)Yield, 88%; mp 219–220°C; 1H-NMR (DMSO-d6, 500 MHz) δ: 4.29 (2H, s), 7.57 (2H, d, J=8.4 Hz), 7.87 (2H, d, J=8.4 Hz), 7.98 (2H, d, J=5.7 Hz), 8.84 (2H, d, J=5.7 Hz), 10.49 (1H, s), 10.65 (1H, s). 13C-NMR (DMSO-d6, 500 MHz) δ: 33.9, 120.6, 128.5, 129.3, 130.9, 131.1, 136.7, 149.8, 163.4, 164.3, 164.8, 165.5. IR (KBr) cm−1: 3352–3221, 3035, 2943, 1705, 1658, 1593. ESI-MS m/z: 390.90 (M+2+·), 388.90 (M+·), 141.00, 139.00, 78.00, 51.00. Anal. Calcd for C16H12ClN5O3S: C, 49.30; H, 3.10; N, 17.97. Found: C, 49.33; H, 3.15; N, 18.14.
4-Ethyl-N′-({[5-(pyridin-4-yl)-1,3,4-oxadiazol-2-yl]sulfanyl}acetyl)benzohydrazide (22)Yield, 92%; mp 195–196°C; 1H-NMR (DMSO-d6, 500 MHz) δ: 1.19 (3H, t, J=7.5, 7.5 Hz), 2.65 (2H, q, J=7.5 Hz), 4.27 (2H, s), 7.31 (2H, d, J=8.4 Hz), 7.78 (2H, d, J=8.4 Hz), 7.94 (2H, d, J=6 Hz), 8.82 (2H, d, J=6 Hz), 10.39 (1H, s), 10.43 (1H, s). IR (KBr) cm−1: 3400–3350, 3071, 2965, 2868, 1677, 1650, 1637. ESI-MS m/z: 383.20 (M+·), 133.15, 105.10, 78.05. Anal. Calcd for C18H17N5O3S: C, 56.38; H, 4.47; N, 18.27. Found: C, 56.36; H, 4.49; N, 18.38.
2-Fluoro-N′-({[5-(pyridin-4-yl)-1,3,4-oxadiazol-2-yl]sulfanyl}acetyl)benzohydrazide (23)Yield, 87%; mp 196–197°C; 1H-NMR (DMSO-d6, 500 MHz) δ: 4.24 (2H, s), 7.25–7.56 (4H, m), 7.99 (2H, d, J=5.4 Hz), 8.82 (2H, d, J=5.4 Hz), 10.40 (1H, s), 10.54 (1H, s). IR (KBr) cm−1: 3250–3201, 3032, 2978, 2885, 1681, 1643, 1612. ESI-MS m/z: 373.25, 220.10, 123.15, 78.10. Anal. Calcd for C16H12FN5O3S: C, 51.47; H, 3.24; N, 18.76. Found: C, 51.58; H, 3.29; N, 18.91.
4-Nitro-N′-({[5-(pyridin-4-yl)-1,3,4-oxadiazol-2-yl]sulfanyl}acetyl)benzohydrazide (24)Yield, 87%; mp 210–211°C; 1H-NMR (DMSO-d6, 500 MHz) δ: 4.30 (2H, s), 7.73–8.84 (8H, m), 10.59 (1H, s), 10.92 (1H, s). IR (KBr) cm−1: 3300–3236, 3005, 2943, 1705, 1654, 1612. ESI-MS m/z: 400.30 (M+·), 399.30, 323.20, 77.10. Anal. Calcd for C16H12N6O5S: C, 48.00; H, 3.02; N, 20.99. Found: C, 48.13; H, 3.08; N, 21.33.
General Procedure for the Preparation of 2-Aryl-5-{[5-(pyridin-4-yl)-1,3,4-oxadiazol-2-ylsulfanyl]methyl}-1,3,4-oxadiazole (25–29)A mixture of 20–23 or 24 (0.001 mol) and phosphorus oxychloride (8 mL) was heated under reflux for 6 h. Excess phosphorus oxychloride was distilled off and the residue was poured onto ice. The separated solid product was filtered, dried and crystallized from aqueous ethanol to give compounds 25–29 respectively.
4-(5-{[(5-Phenyl-1,3,4-oxadiazol-2-yl)methyl]sulfanyl}-1,3,4-oxadiazol-2-yl)pyridine (25)Yield, 42%; mp 150–151°C; 1H-NMR (CDCl3, 500 MHz) δ: 4.84 (2H, s), 7.50–8.90 (9H, m). IR (KBr) cm−1: 3066, 2924, 2854, 1600. ESI-MS m/z: 337.05 (M+·), 105.05, 106.05, 78.05, 77.05, 51.05. Anal. Calcd for C16H11N5O2S: C, 56.96; H, 3.29; N, 20.76. Found: C, 57.11; H, 3.32; N, 20.89.
4-[5-({[5-(4-Chlorophenyl)-1,3,4-oxadiazol-2-yl]methyl}sulfanyl)-1,3,4-oxadiazol-2-yl]pyridine (26)Yield, 63%; mp 190–191°C; 1H-NMR (CDCl3, 500 MHz) δ: 4.83 (2H, s), 7.48 (2H, d, J=9 Hz), 7.97 (2H, d, J=9 Hz), 8.20 (2H, d, J=5.4 Hz), 8.89 (2H, d, J=5.4 Hz). IR (KBr) cm−1: 3050, 2924, 2854, 1593. ESI-MS m/z: 373.10 (M+2+·), 371.10 (M+·), 78.10, 51.05. Anal. Calcd for C16H10ClN5O2S: C, 51.69; H, 2.71; N, 18.84. Found: C, 51.76; H, 2.74; N, 19.12.
4-[5-({[5-(4-Ethylphenyl)-1,3,4-oxadiazol-2-yl]methyl}sulfanyl)-1,3,4-oxadiazol-2-yl]pyridine (27)Yield, 63%; mp 129–130°C; 1H-NMR (DMSO-d6, 500 MHz) δ: 1.22 (3H, t, J=7.5, 7.5 Hz), 2.60 (2H, q, J=7.5 Hz), 4.98 (2H, s), 7.40–8.81 (8H, m). IR (KBr) cm−1: 3090, 2958, 1603. ESI-MS m/z: 365.20 (M+·), 149.05. Anal. Calcd for C18H15N5O2S: C, 59.16; H, 4.14; N, 19.17. Found: C, 59.18; H, 4.19; N, 19.36.
4-[5-({[5-(2-Fluorophenyl)-1,3,4-oxadiazol-2-yl]methyl}sulfanyl)-1,3,4-oxadiazol-2-yl]pyridine (28)Yield, 58%; mp 139–140°C; 1H-NMR (CDCl3, 500 MHz) δ: 4.03 (2H, s), 7.33–8.76 (8H, m). 13C-NMR (DMSO-d6, 500 MHz) δ: 30.6, 116.4, 116.7, 120.7, 121.8, 124.8, 130.2, 134.0, 141.8, 149.3, 157.7, 162.3, 166.3, 168.8. IR (KBr) cm−1: 3070, 2850, 1620. ESI-MS m/z: 355.25 (M+·), 123.15, 106.00, 95.10, 78.10, 51.05. Anal. Calcd for C16H10FN5O2S: C, C 54.08; H, 2.84; N, 19.71. Found: C, 54.13; H, 2.89; N, 19.81.
4-[5-({[5-(4-Nitrophenyl)-1,3,4-oxadiazol-2-yl]methyl}sulfanyl)-1,3,4-oxadiazol-2-yl]pyridine (29)Yield, 61%; mp 180–181°C; 1H-NMR (CDCl3, 500 MHz) δ: 4.11 (2H, s), 6.92–8.83 (8H, m). IR (KBr) cm−1: 3050, 2924, 2854, 1597. ESI-MS m/z: 382.00 (M+·), 150.00, 146.00, 106.00, 78.00, 51.00. Anal. Calcd for C16H10N6O4S: C, 50.26; H, 2.64; N, 21.98. Found: C, 50.32; H, 2.71; N, 22.12.
General Procedure for the Preparation of N-Aryl-2-{2-[5-(pyridin-4-yl)-1,3,4-oxadiazol-2-ylsulphanyl]acetyl}hydrazinecarbothioamides (30–32)A mixture of equimolar amounts of the hydrazide 11 (0.25 g, 0.001 mol) and the appropriate phenyl isothiocyanate (0.001 mol) in absolute ethanol (15 mL) was heated under reflux for 6 h. After cooling, the precipitated solid was filtered, dried and crystallized from DMF/ethanol to give compounds 30–32.
N-Phenyl-2-({[5-(pyridin-4-yl)-1,3,4-oxadiazol-2-yl]sulfanyl}acetyl)hydrazinecarbothioamide (30)Yield, 52%; mp 234–235°C; 1H-NMR (DMSO-d6, 500 MHz) δ: 4.25 (2H, s), 7.17–7.44 (5H, m), 7.90 (2H, d, J=5.7 Hz), 8.81 (2H, d, J=5.7 Hz), 9.50 (1H, s), 9.76 (1H, s), 10.46 (1H, s). IR (KBr) cm−1: 3390–3174, 3055, 2966, 2924, 1681, 1600, 1234. ESI-MS m/z: 386.50 (M+·), 250.40. Anal. Calcd for C16H14N6O2S2: C, 49.73; H, 3.65; N, 21.75. Found: C, 49.78; H, 3.63; N, 21.82.
N-(4-Chlorophenyl)-2-({[5-(pyridin-4-yl)-1,3,4-oxadiazol-2-yl]sulfanyl}acetyl)hydrazine Carbothioamide (31)Yield, 69%; mp 215–216°C; 1H-NMR (DMSO-d6, 500 MHz) δ: 4.22 (2H, s), 7.35 (2H, d, J=8.4 Hz), 7.52–7.65 (2H, m), 7.87 (2H, d, J=5.4 Hz), 8.77 (2H, d, J=5.4 Hz), 9.73 (1H, s), 9.95 (1H, s), 10.48 (1H, s). IR (KBr) cm−1: 3278–3194, 3074, 2850, 1681, 1612, 1234. ESI-MS m/z: 422.25 (M+2+·), 420.20 (M+·), 129.00, 127.00, 113.00, 111.00, 78.00, 51.00. Anal. Calcd for C16H13ClN6O2S2: C, 45.66; H, 3.11; N, 19.97. Found: C, 45.72; H, 3.18; N, 20.08.
N-(4-Methoxyphenyl)-2-({[5-(pyridin-4-yl)-1,3,4-oxadiazol-2-yl]sulfanyl}acetyl)hydrazine Carbothioamide (32)Yield, 65%; mp 207–208°C. 1H-NMR (DMSO-d6, 500 MHz) δ: 3.71 (3H, s), 4.21 (2H, s), 6.85–7.35 (4H, m), 7.87 (2H, d, J=4.5 Hz), 8.77 (2H, d, J=4.5 Hz), 9.50 (1H, s), 9.68 (1H, s), 10.43 (1H, s). IR (KBr) cm−1: 3205–3190, 3070, 2958, 2835, 1678, 1604, 1246. ESI-MS m/z: 416.00 (M+·), 250.00, 92.00, 78.00, 77.00. Anal. Calcd for C17H16N6O3S2: C, 49.03; H, 3.87; N, 20.18. Found: C, 49.13; H, 3.92; N, 20.45.
General Procedure for the Preparation of N′-{2-[5-(Pyridin-4-yl)-1,3,4-oxadiazol-2-ylsulphanyl]acetyl}arylsulphonohydrazides (33, 34)To a solution of 33 (0.50 g, 0.002 mol) in dry pyridine (10 mL), the appropriate aryl sulphonyl chloride (0.002 mol) was added. The reaction mixture was stirred at room temperature for 5 h, and then poured onto ice-cold water. The formed precipitate was filtered, dried and crystallized from ethanol to give compounds 33 and 34.
N′-(Phenylsulfonyl)-2-{[5-(pyridin-4-yl)-1,3,4-oxadiazol-2-yl]sulfanyl}acetohydrazide (33)Yield, 50%; mp 180–181°C; 1H-NMR (DMSO-d6, 500 MHz) δ: 4.04 (2H, s), 7.45–7.78 (5H, m), 7.90 (2 H, d, J=4.5 Hz), 8.83 (2H, d, J=4.5 Hz), 10.04 (1H, d, J=3.3 Hz), 10.45 (1H, d, J=3.3 Hz). IR (KBr) cm−1: 3433–3186, 3071, 2848, 1663, 1575, 1304, 1158. ESI-MS m/z: 391.20 (M+·), 51.00. Anal. Calcd for C15H13N5O4S2: C, 46.03; H, 3.35; N, 17.89. Found: C, 46.09; H, 3.37; N, 18.02.
N′-[(4-Methylphenyl)sulfonyl]-2-{[5-(pyridin-4-yl)-1,3,4-oxadiazol-2-yl]sulfanyl}acetohydrazide (34)Yield, 62%; mp 190–191°C; 1H-NMR (DMSO-d6, 500 MHz) δ: 2.29 (3H, s), 4.03 (2H, s), 7.26 (2H, d, J=8 Hz), 7.64 (2H, d, J=8 Hz), 7.90 (2H, d, J=6 Hz), 8.83 (2H, d, J=6 Hz), 9.93 (1H, d, J=3.3 Hz), 10.41 (1H, d, J=3.3 Hz). 13C-NMR (DMSO-d6, 500 MHz) δ: 20.9, 33.5, 119.9, 120.6, 127.5, 129.2, 135.9, 143.1, 150.9, 163.5, 164.2, 165.1. IR (KBr) cm−1: 3425–3199, 3076, 2984, 2849, 1669, 1606, 1344, 1161. ESI-MS m/z: 405.10 (M+·), 250.10, 220.05. Anal. Calcd for C16H15N5O4S2: C, 47.40; H, 3.73; N, 17.27. Found: C, 47.00; H, 4.00; N, 17.50.
Potassium 2-{2-[5-(Pyridin-4-yl)-1,3,4-oxadiazol-2-ylsulfanyl]acetyl}hydrazinecarbodithioate (35)To a solution of 11 (1.00 g, 0.004 mol) and potassium hydroxide (0.34 g, 0.006 mol) in ethanol (15 mL), carbon disulfide (0.38 g, 0.005 mol) was added portion wise with stirring and cooling in ice bath. The stirring was continued for 12 h at room temperature; the precipitated solid was filtered, washed with ether and dried to give compound 35.
Yield, 52%; mp 234–235°C. 1H-NMR (DMSO-d6, 300 MHz) δ: 3.72 (2H, s), 5.21 (2H, s), 7.80 (2H, d, J=5.7 Hz), 8.47 (2H, d, J=5.7 Hz). ; IR (KBr) cm−1: 3317–3213, 3062, 2954, 1693, 1230. ESI-MS m/z: 365.10 (M+·), 219.00, 68.00. Anal. Calcd for C10H8KN5O2S3, Calcd. C, 32.86; H, 2.21; N, 19.16, Found. C, 32.91; H, 2.23; N, 19.24.
4-Amino-5-{[4-amino-5-(pyridin-4-yl)-4H-1,2,4-triazol-3-ylsulfanyl]methyl}-4H-1,2,4-triazole-3-thiol (36)To a solution of potassium dithiocarbazinate 35 (1.10 g, 0.003 mol) in water (10 mL), hydrazine hydrate (99%, 0.30 g, 0.006 mol) was added, and the reaction mixture was heated under reflux for 20 h. After cooling, the reaction mixture was diluted with ice-cold water (10 mL) and acidified with acetic acid (1 mL). The formed precipitate was filtered, dried then crystallized from ethanol to give compound 36.
Yield, 46%; mp 230–231°C; 1H-NMR (DMSO-d6, 500 MHz) δ: 4.01 (2H, s), 5.82 (2H, s), 6.28 (2H, s), 7.96 (2H, d, J=5 Hz), 8.70 (2H, d, J=5 Hz), 14.14 (1H, s). IR (KBr) cm−1: 3286–3174, 2542, 1627. ESI-MS m/z: 321.05 (M+·), 192.95, 78.00, 51.00. Anal. Calcd for C10H11N9S2, Calcd. C, 37.37; H, 3.45; N, 39.22, Found, C, 37.41; H, 3.42; N, 39.45.
Computer-Aided Prediction of Biological ActivityThe five parameters proposed by Lipinski for prediction of intestinal absorption and blood brain barrier penetration (molecular weight (MW), NROTB, HBA, hydrogen bond donors (HBD) and Log P). These parameters were calculated for all target compounds using Molinspiration software (Molsoft 2007) http://www.molinspiration.com/cgi-bin/properties. The calculated values of the newly synthesized compounds were compared with that of erlotinib.
Cytotoxic ActivityIn this study twenty eight newly synthesized compounds out of thirty six were tested for their anticancer activity against breast carcinoma cell line (MCF-7) at Pharmacology Lab, Cancer Biology Unit at the Egyptian National Cancer Institute. Sulphorhodamine-B (SRB) assay method was carried out according to that of Monk et al.25) Cells were plated in 96-multiwell plate (104 cells/well) for 24h before treatment with tested compounds to allow attachment of cells to the wall of the plate. Different concentrations of each compound (0, 5, 12.5, 25 and 50 µg/mL) were added to the cell monolayer triplicate wells prepared for each individual dose. Monolayer cells were incubated with tested compounds for 48 h at 37°C and in atmosphere of 5% CO2. After 48 h cells were fixed, washed and stained with sulphorhodamine-B stain. Excess stain was washed with 1% acetic acid and color intensity was measured spectrophotometrically with an enzyme-linked immunosorbent assay (ELISA) reader. The relation between surviving fraction and drug concentration was plotted to get the survival curve and IC50 was calculated for each compound and compared with reference drug Erlotinib with IC50=0.020 µM, each concentration was repeated 3 times for each compound.
Docking StudiesThe X-ray crystallographic structure of the active site of EGFR (PDB ID: 1M17) was obtained from protein data bank available at the RCSB Protein Data Bank, (http://www.rcsb.org).
Compounds were built using Molecular Operating Environment (MOE) molecule builder. The structures were energy minimized by Merck Molecular force field (MMff 94x). Hydrogen atoms and partial charges were added to the system using protonate 3D application. Compounds were grouped in databases, docked using the MOE Dock tool and default settings were used. Poses were generated by superposition of ligand atom triplets and triplets of points in the receptor binding site in a systematic way. Poses generated by the placement methodology were scored using an available method implemented in MOE. Poses resulting from the placement stage were then subjected to MMFF94x energy minimization. For each docked compound only one pose was selected based on number of binding interactions, superposition with the original ligand, docking score and the formed H-bonds lengths.
The authors are grateful to Prof. Dr. Samia Shouman, Professor of Pharmacology, and all members of the Cancer Biology Department, National Cancer Institute, Cairo, Egypt, for carrying out the cytotoxicity testing.
The authors declare no conflict of interest.