Abstract
Irciniastatin A is a pederin-type marine product that potently inhibits translation. We have recently shown that irciniastatin A induces ectodomain shedding of tumor necrosis factor (TNF) receptor 1 with slower kinetics than other translation inhibitors. In human lung carcinoma A549 cells, irciniastatin A induced a marked and sustained activation of extracellular signal-regulated kinase (ERK) and induced little activation of p38 mitogen-activated protein (MAP) kinase and c-Jun N-terminal kinase (JNK). Moreover, the TNF receptor 1 shedding induced by irciniastatin A was blocked by the MAP kinase/ERK kinase inhibitor U0126, but not by the p38 MAP kinase inhibitor SB203580 or the JNK inhibitor SP600125. Thus unlike other translation inhibitors that trigger ribotoxic stress response, our results show that irciniastatin A is a unique translation inhibitor that induces a potent and sustained activation of the ERK pathway, and thereby promotes the ectodomain shedding of TNF receptor 1 in A549 cells.
Irciniastatin A, also known as psymberin (Fig. 1A), is a marine natural product originally isolated from the marine sponges Ircinia ramose and Psammocinia.1,2) Irciniastatin A belongs to the pederin family, including mycalamides, and exerts potent antitumor activity.3) It has been reported that pederin-type compounds inhibit protein synthesis,4–6) and mycalamide A and B have been shown to bind directly to the E site of the large ribosomal subunit.6,7) In previous reports, we synthesized irciniastatin A and observed that it potently inhibits protein synthesis.8,9) Additionally, genetic studies with Caenorhabditis elegans show that irciniastatin A primarily targets the ribosome.10)
Cell-surface tumor necrosis factor (TNF) receptor 1 is processed into its soluble form by TNF-α-converting enzyme (TACE), also referred to as a disintegrin and metalloproteinase 17.11) We have previously shown that three translation inhibitors, i.e., acetoxycycloheximide, cytotrienin A and deoxynivalenol, rapidly induced the ectodomain shedding of TNF receptor 1 in human lung carcinoma A549 cells.12–14) Moreover, we have recently reported that irciniastatin A induced the ectodomain shedding of TNF receptor 1 with slower kinetics than the translation inhibitors.15)
Some types of translation inhibitors are known to trigger ribotoxic stress response.16) The ribotoxic stress response leads to the activation of mitogen-activated protein (MAP) kinase superfamily members, extracellular signal-regulated kinase (ERK), p38 MAP kinase, and c-Jun N-terminal kinase (JNK); these kinases regulate various intracellular signaling pathways and cellular processes.17,18) In particular, ERK and p38 MAP kinase phosphorylate TACE at Thr-735 in the cytoplasmic domain, and regulate the TACE-dependent ectodomain shedding.19–21) We have reported that acetoxycycloheximide and deoxynivalenol induce a rapid activation of ERK and p38 MAP kinase, and thereby promote the ectodomain shedding of TNF receptor 1 in A549 cells.14,22) In this study, we further investigated the mechanism by which irciniastatin A promotes the ectodomain shedding of TNF receptor 1.
MATERIALS AND METHODS
CellsHuman lung carcinoma A549 cells (JCRB0076) were provided by the National Institute of Biomedical Innovation JCRB Bank (Osaka, Japan). A549 cells were maintained in RPMI 1640 medium (Invitrogen, Carlsbad, CA, U.S.A.), supplemented with heat-inactivated fetal calf serum (Nichirei Biosciences, Tokyo, Japan) and penicillin–streptomycin solution (Nacalai Tesque, Kyoto, Japan).
ReagentsIrciniastatin A was prepared as described previously.8) Deoxynivalenol and SP600125 were obtained from Sigma-Aldrich (St. Louis, MO, U.S.A.). Puromycin (Nacalai Tesque), SB203580 (Cayman Chemical, Ann Arbor, MI, U.S.A.), and U0126 (Wako Pure Chemical Industries, Ltd., Osaka, Japan) were obtained commercially.
Preparation of Cell LysatesCulture media were collected by centrifugation (800×g, 5 min). Proteins were precipitated by chloroform/methanol. Cells were washed once with phosphate-buffered saline and lysed with Triton X-100 lysis buffer consisting of 50 mM Tris–HCl (pH 7.4), 1% Triton X-100, 2 mM dithiothreitol and protease inhibitor cocktail (Complete™; Roche Diagnostics, Mannheim, Germany). Cell lysates were centrifuged (15300×g, 5 min) and supernatants were collected as cytoplasmic fractions. For the preparation of whole cell lysates, cells were lysed with Triton X-100 lysis buffer containing phosphatase inhibitor cocktail (Nacalai Tesque), followed by sonication and centrifugation (15300×g, 5 min).
Western BlottingProtein samples (30 µg) were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred onto nitrocellulose membranes. Primary antibodies specific to β-actin (AC-15; Sigma-Aldrich), ERK1/ERK2 (#9102; Cell Signaling Technology, Danvers, MA, U.S.A.), JNK (#9252; Cell Signaling Technology), p38 MAP kinase (#9212; Cell Signaling Technology), phospho-ERK1/ERK2 (Thr202/Tyr204) (#9101; Cell Signaling Technology), phospho-JNK (Thr183/Tyr185) (#9251; Cell Signaling Technology), phospho-p38 MAP kinase (Thr180/Tyr182) (#9211; Cell Signaling Technology), TNF receptor 1 (H-5; Santa Cruz Biotechnology, Santa Cruz, CA, U.S.A.) and horseradish peroxidase-linked secondary antibodies (Jackson ImmunoResearch Laboratories, West Grove, PA, U.S.A.) were used for Western blotting. The single membrane was re-probed for different proteins or the multiple membranes derived from the same samples were used for Western blotting using different antibodies. Blots were analyzed by an ImageQuant LAS 4000 Mini (GE Healthcare, Piscataway, NJ, U.S.A.).
Statistical AnalysisStatistical significance was evaluated by one-way ANOVA followed by the Tukey test for multiple comparisons. Differences with p<0.05 were considered to be statistically significant.
RESULTS AND DISCUSSION
We compared the effect of two translation inhibitors, irciniastatin A, and puromycin, on the expression of TNF receptor 1 in A549 cells. As recently reported,15) irciniastatin A increased the amount of soluble TNF receptor 1 in the culture medium for 3 h (Fig. 1B). Under the same incubation period, puromycin did not significantly increase the amount of soluble TNF receptor 1 (Fig. 1B). This is consistent with the fact that puromycin does not induce ribotoxic stress response.5,16,23) Both irciniastatin A and puromycin similarly reduced the amount of total TNF receptor 1 in the cell lysates (Fig. 1B). Thus, it seems likely that cell-surface TNF receptor 1 is processed into its soluble form via the ectodomain shedding induced by irciniastatin A, while intracellular TNF receptor 1 is reduced mainly by inhibiting the translation process in irciniastatin-A- or puromycin-treated cells.
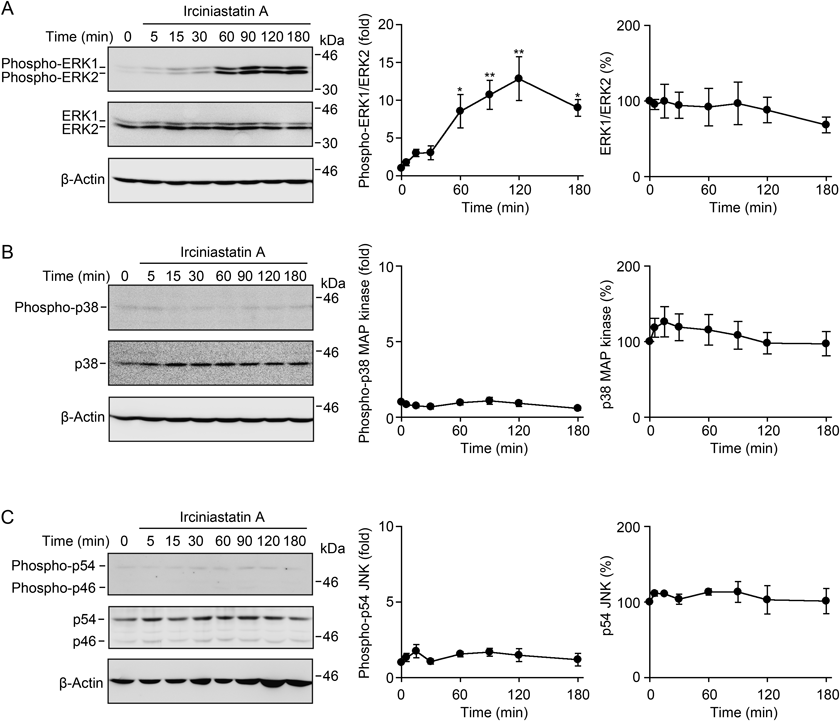
We have previously shown that both ERK and p38 MAP kinase are responsible for the ectodomain shedding of TNF receptor 1 in A549 cells when treated with acetoxycycloheximide, cytotrienin A, and deoxynivalenol.13,14,22) We analyzed the activation of the ERK, p38 MAP kinase and JNK pathways by Western blotting using antibodies specific for phospho-ERK, phospho-p38 MAP kinase and phospho-JNK. Two ERK isoforms, ERK1 and ERK2, were gradually phosphorylated starting 30 min after exposure to irciniastatin A, and the phosphorylation states of ERK1 and ERK2 were maintained at a high level between 60–180 min in irciniastatin-A-treated A549 cells (Fig. 2A). By contrast, p38 MAP kinase and JNK were barely phosphorylated up to 180 min after exposure to irciniastatin A (Figs. 2B, C).
Deoxynivalenol is known to induce the activation of ERK, p38 MAP kinase and JNK.14,24–26) As shown previously in A549 cells,14) deoxynivalenol increased transient phosphorylation of ERK1 and ERK2 at 15 min (Fig. 3A). We observed that irciniastatin A induced the phosphorylation of ERK1 and ERK2 at levels much higher than those induced by deoxynivalenol (Fig. 3A). By contrast, irciniastatin A barely induced phosphorylation of p38 MAP kinase and JNK at either 15 min or 120 min, despite being under the same conditions in which deoxynivalenol was able to significantly induce phosphorylation of p38 MAP kinase and JNK (Figs. 3B, C). These data indicate that irciniastatin A induces the potent and sustained activation of ERK, but little activation of p38 MAP kinase and JNK in A549 cells.
To identify the protein kinase(s) responsible for TNF receptor 1 shedding, A549 cells were preincubated with specific protein kinase inhibitors for 1 h and then incubated with irciniastatin A for 3 h. The MAP kinase/ERK kinase (MEK) inhibitor U0126 diminished the increased shedding of TNF receptor 1 by irciniastatin A, as well as the constitutive shedding of TNF receptor 1 (Fig. 4A). However, neither the p38 MAP kinase inhibitor SB203580 nor the JNK inhibitor SP600125 affected the ectodomain shedding of TNF receptor 1 in irciniastatin-A-treated cells (Fig. 4A). The phosphorylation of ERK1/ERK2 induced by irciniastatin A was markedly inhibited by U0126, but not by SB203580 or SP600125 (Fig. 4B). None of the three kinase inhibitors suppressed the decrease of total TNF receptor 1 in irciniastatin-A-treated cells (Fig. 4A). Thus, these data indicate that the ERK pathway, but neither the p38 MAP kinase pathway nor the JNK pathway, is responsible for the ectodomain shedding of TNF receptor 1 induced by irciniastatin A, unlike other translation inhibitors. This is also supported by our previous observation that the ectodomain shedding of TNF receptor 1 induced by deoxynivalenol was inhibited partially by either U0126 or SB20350, but completely by the combination of U0126 and SB203580.14)
ERK and p38 MAP kinase regulate the TACE-mediated ectodomain shedding of cell-surface proteins.19–21,27) Consistent with this notion, we have previously shown that both ERK and p38 MAP kinase are responsible for the TNF receptor 1 shedding induced by acetoxycycloheximide, cytotrienin A and deoxynivalenol in A549 cells.13,14,22) However, unlike these translation inhibitors, our results revealed that only ERK is responsible for the TNF receptor 1 shedding induced by irciniastatin A. In particular, irciniastatin A induced ERK activation with slower kinetics, yet much more potently compared with deoxynivalenol. Thus, irciniastatin-A-induced ERK activation may have a mechanism quite different from other translation inhibitors capable of inducing ribotoxic stress response.
Irciniastatin A barely promoted the phosphorylation of p38 MAP kinase and JNK in A549 cells. By contrast, we have previously shown that irciniastatin A increases the phosphorylation of p38 MAP kinase and JNK in human leukemia Jurkat cells.9) These results suggest that irciniastatin A may induce the activation of p38 MAP kinase and JNK in a cell-type-specific manner. This seems to be a unique property of irciniastatin A, because we have previously reported that acetoxycycloheximide is able to induce the activation of p38 MAP kinase and JNK in both A549 cells and Jurkat cells.22,28) The ectopic expression of the Bcl-2 family member Bcl-xL or the reactive oxygen species scavenger N-acetyl-L-cysteine diminished the increased phosphorylation of p38 MAP kinase and JNK in irciniastatin-A-treated Jurkat cells.9) These results imply that the activation of p38 MAP kinase and JNK by irciniastatin A may be mediated via the mitochondrial pathway. Further clarification is necessary to understand the molecular mechanism by which irciniastatin A induces the activation of ERK, p38 MAP kinase and/or JNK in different cell types. In conclusion, the present study indicates that irciniastatin A is a unique translation inhibitor that induces the potent and sustained activation of the ERK pathway in A549 cells.
Acknowledgments
This work was supported by a Grant-in-Aid for Scientific Research (KAKENHI, Grant Numbers 22380060 and 25292061 to T.K.) from the Japan Society for the Promotion of Science (JSPS) and a Grant-in-Aid for Scientific Research on Innovated Areas “Chemical Biology of Natural Products” (to N.K. and T.U.) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan.
Conflict of Interest
The authors declare no conflict of interest.
REFERENCES
- 1) Cichewicz RH, Valeriote FA, Crews P. Psymberin, a potent sponge-derived cytotoxin from Psammocinia distantly related to the pederin family. Org. Lett., 6, 1951–1954 (2004).
- 2) Pettit GR, Xu JP, Chapuis JC, Pettit RK, Tackett LP, Doubek DL, Hooper JNA, Schmidt JM. Antineoplastic agents. 520. Isolation and structure of irciniastatins A and B from the Indo-Pacific marine sponge Ircinia ramose. J. Med. Chem., 47, 1149–1152 (2004).
- 3) Bielitza M, Pietruszka J. The psymberin story—biological properties and approaches towards total and analogue syntheses. Angew. Chem. Int. Ed., 52, 10960–10985 (2013).
- 4) Burres NS, Clement JJ. Antitumor activity and mechanism of action of the novel marine natural products mycalamide-A and -B and onnamide. Cancer Res., 49, 2935–2940 (1989).
- 5) Lee KH, Nishimura S, Matsunaga S, Fusetani N, Horinouchi S, Yoshida M. Inhibition of protein synthesis and activation of stress-activated protein kinases by onnamide A and theopederin B, antitumor marine natural products. Cancer Sci., 96, 357–364 (2005).
- 6) Dang Y, Schneider-Poetsch T, Eyler DE, Jewett JC, Bhat S, Rawal VH, Green R, Liu JO. Inhibition of eukaryotic translation elongation by the antitumor natural product mycalamide B. RNA, 17, 1578–1588 (2011).
- 7) Gürel G, Blaha G, Steitz TA, Moore PB. Structures of triacetyloleandomycin and mycalamide A bind to the large ribosome subunit of Haloarcula marismortui. Antimicrob. Agents Chemother., 53, 5010–5014 (2009).
- 8) Watanabe T, Imaizumi T, Chinen T, Nagumo Y, Shibuya M, Usui T, Kanoh N, Iwabuchi Y. Syntheses and biological evaluation of irciniastatin A and the C1–C2 alkyne analogue. Org. Lett., 12, 1040–1043 (2010).
- 9) Chinen T, Nagumo Y, Watanabe T, Imaizumi T, Shibuya M, Kataoka T, Kanoh N, Iwabuchi Y, Usui T. Irciniastatin A induces JNK activation that is involved in caspase-8-dependent apoptosis via the mitochondrial pathway. Toxicol. Lett., 199, 341–346 (2010).
- 10) Wu CY, Feng Y, Cardenas ER, Williams N, Floreancig PE, De Brabander JK, Roth MG. Studies toward the unique pederin family member psymberin: structure–activity relationships, biochemical studies, and genetics identify the mode-of-action of psymberin. J. Am. Chem. Soc., 134, 18998–19003 (2012).
- 11) Scheller J, Chalaris A, Garbers C, Rose-John S. ADAM17: a molecular switch to control inflammation and tissue regeneration. Trends Immunol., 32, 380–387 (2011).
- 12) Ogura H, Tsukumo Y, Sugimoto H, Igarashi M, Nagai K, Kataoka T. Ectodomain shedding of TNF receptor 1 induced by protein synthesis inhibitors regulates TNF-α-mediated activation of NF-κB and caspase-8. Exp. Cell Res., 314, 1406–1414 (2008).
- 13) Yamada Y, Taketani S, Osada H, Kataoka T. Cytotrienin A, a translation inhibitor that induces ectodomain shedding of TNF receptor 1 via activation of ERK and p38 MAP kinase. Eur. J. Pharmacol., 667, 113–119 (2011).
- 14) Hirano S, Kataoka T. Deoxynivalenol induces ectodomain shedding of TNF receptor 1 and thereby inhibits the TNF-α-induced NF-κB signaling pathway. Eur. J. Pharmacol., 701, 144–151 (2013).
- 15) Hirano S, Quach HT, Watanabe T, Kanoh N, Iwabuchi Y, Usui T, Kataoka T. Irciniastatin A, a pederin-type translation inhibitor, promotes ectodomain shedding of cell-surface tumor necrosis factor receptor 1. J. Antibiot. (Tokyo), (2015), in press.
- 16) Iordanov MS, Pribnow D, Magun JL, Dinh TH, Pearson JA, Chen SLY, Magun BE. Ribotoxic stress response: activation of the stress-activated protein kinase JNK1 by inhibitors of the peptidyl transferase reaction and by sequence-specific RNA damage to the α-sarcin/ricin loop in the 28S rRNA. Mol. Cell. Biol., 17, 3373–3381 (1997).
- 17) Pestka JJ. Deoxynivalenol: mechanism of action, human exposure, and toxicological relevance. Arch. Toxicol., 84, 663–679 (2010).
- 18) Kataoka T. Translation inhibitors and their unique biological properties. Eur. J. Pharmacol., 676, 1–5 (2012).
- 19) Díaz-Rodríguez E, Montero JC, Esparís-Ogando A, Yuste L, Pandiella A. Extracellular signal-regulated kinase phosphorylates tumor necrosis factor α-converting enzyme at threonine 735: a potential role in regulated shedding. Mol. Biol. Cell, 13, 2031–2044 (2002).
- 20) Soond SM, Everson B, Riches DWH, Murphy G. ERK-mediated phosphorylation of Thr735 in TNFα-converting enzyme and its potential role in TACE protein trafficking. J. Cell Sci., 118, 2371–2380 (2005).
- 21) Xu P, Derynck R. Direct activation of TACE-mediated ectodomain shedding by p38 MAP kinase regulates EGF receptor-dependent cell proliferation. Mol. Cell, 37, 551–566 (2010).
- 22) Ogura H, Tsukumo Y, Sugimoto H, Igarashi M, Nagai K, Kataoka T. ERK and p38 MAP kinase are involved in downregulation of cell surface TNF receptor 1 induced by acetoxycycloheximide. Int. Immunopharmacol., 8, 922–926 (2008).
- 23) Sidhu JS, Omiecinski CJ. Protein synthesis inhibitors exhibit a nonspecific effect on phenobarbital-inducible cytochrome P450 gene expression in primary rat hepatocytes. J. Biol. Chem., 273, 4769–4775 (1998).
- 24) Shifrin VI, Anderson P. Trichothecene mycotoxins trigger a ribotoxic stress response that activates c-Jun N-terminal kinase and p38 mitogen-activated protein kinase and induces apoptosis. J. Biol. Chem., 274, 13985–13992 (1999).
- 25) Chung YJ, Zhou HR, Pestka JJ. Transcriptional and posttranslational roles for p38 mitogen-activated protein kinase in upregulation of TNF-α expression by deoxynivalenol (vomitoxin). Toxicol. Appl. Pharmacol., 193, 188–201 (2003).
- 26) Islam Z, Gray JS, Pestka JJ. p38 mitogen-activated protein kinase mediates IL-8 induction by the ribotoxin deoxynivalenol in human monocytes. Toxicol. Appl. Pharmacol., 213, 235–244 (2006).
- 27) Liu C, Xu P, Lamouille S, Xu J, Derynck R. TACE-mediated ectodomain shedding of the type I TGF-β receptor downregulates TGF-β signaling. Mol. Cell, 35, 26–36 (2009).
- 28) Kadohara K, Tsukumo Y, Sugimoto H, Igarashi M, Nagai K, Kataoka T. Acetoxycycloheximide (E-73) rapidly induces apoptosis mediated by the release of cytochrome c via activation of c-Jun N-terminal kinase. Biochem. Pharmacol., 69, 551–560 (2005).